| Size | Price | Stock | Qty |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
Razuprotafib (AKB-9778) is a novel small molecule Tie2 activator being developed in phase 2 clnical trials by Aerpio company for a number of indications including Diabetic Nephropathy, Open Angle Glaucoma and ARDS due to COVID-19. It enhances endothelial function and stabilizes blood vessels, including pulmonary and renal vasculature. Razuprotafib restores Tie2 activation to stabilize the vasculature providing breakthrough potential for reducing the severity of COVID-19 associated pulmonary and vascular pathology resulting in fewer patients requiring ventilator support, decreased time in ICU on ventilator support and more rapid and complete recovery with concomitant reduction mortality. Additionally, razuprotafib together with emerging antiviral drugs could provide the optimal combination of host and virus targeted therapy for prevention and treatment of COVID‑19 and COVID-19 related ARDS. Razuprotafib is also a protein tyrosine phosphatase ß (HPTPß) inhibitor.
Razuprotafib (AKB-9778) is a potent and selective small‑molecule inhibitor of vascular endothelial‑protein tyrosine phosphatase (VE‑PTP), a negative regulator of TIE2 receptor activation. It was developed to stabilize retinal and choroidal vasculature by enhancing TIE2 signaling. In preclinical ocular disease models, Razuprotafib suppresses neovascularization (NV) and vascular leakage, showing potential for treating neovascular age‑related macular degeneration (AMD), diabetic retinopathy, and retinal vein occlusion. [1]| Targets |
Razuprotafib (AKB-9778) targets VE‑PTP (IC50 = 17 pM). It also inhibits the structurally related phosphatases HPTPh (IC50 = 36 pM) and HPTPγ (IC50 = 100 pM). It shows weak inhibition of LAR (IC50 = 295 nM), PTP1B (IC50 = 780 nM), CD45 (IC50 = 7,812 nM), and HPTPe (IC50 = 14,609 nM), while no significant inhibition (IC50 >20,000 nM) is observed against HCPTPA, PRL3, MKP‑1, VHR, ALP, and PP1γ. [1]
|
|---|---|
| ln Vitro |
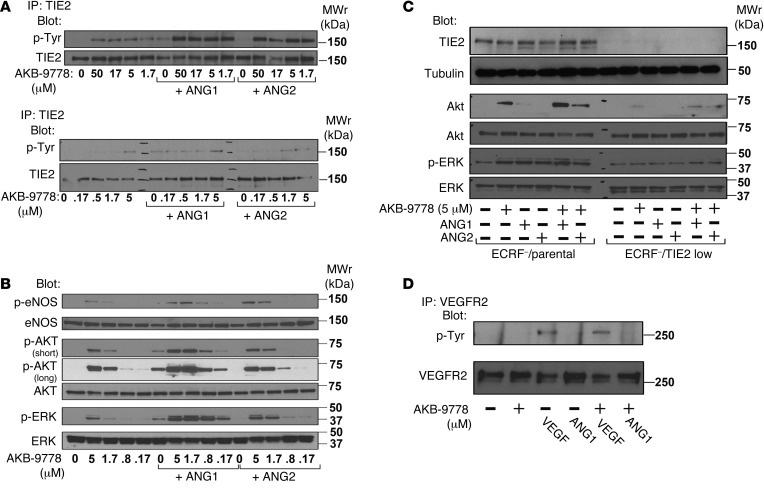
Razuprotafib (AKB-9778) increases angiopoietin-induced TIE2 phosphorylation and stimulates TIE2 phosphorylation and downstream signaling in HUVECs [1].
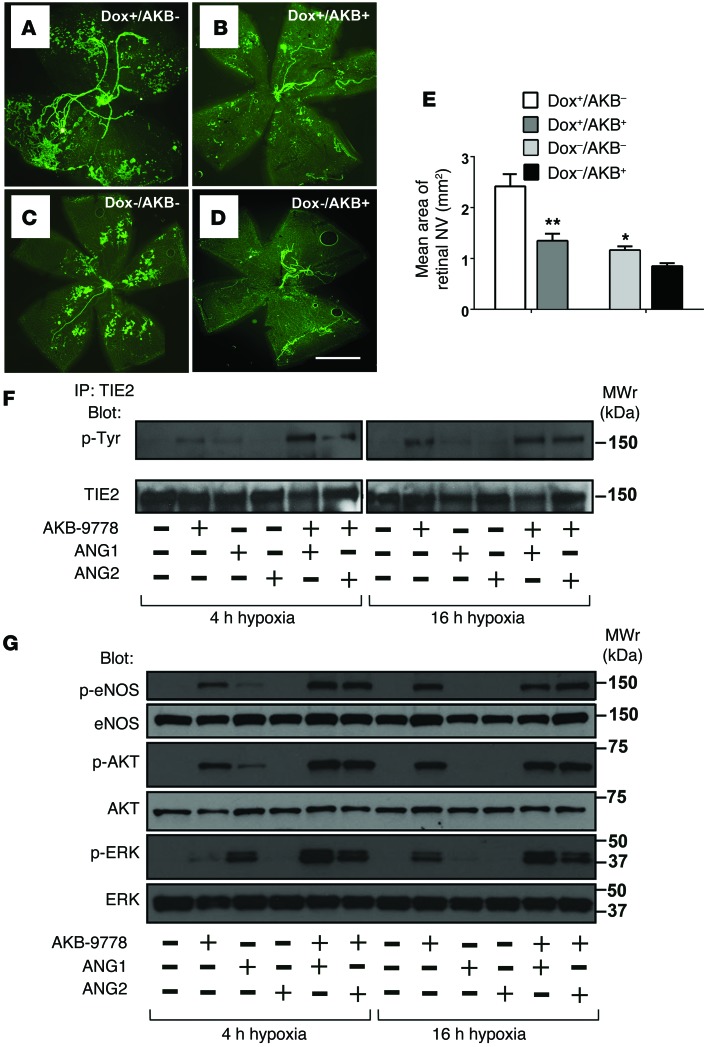
In HUVECs, Razuprotafib (AKB-9778) induces concentration‑dependent TIE2 tyrosine phosphorylation at 0.17–50 μM. [1] At 1.7 μM, it causes TIE2, AKT, eNOS, and ERK phosphorylation comparable to 500 ng/mL ANG1. Co‑incubation with ANG1 or ANG2 markedly enhances these effects. [1] Knockdown of TIE2 by shRNA abrogates Razuprotafib‑induced AKT and ERK phosphorylation, confirming target specificity. [1] Razuprotafib does not induce VEGFR2 phosphorylation nor affect VEGF‑induced VEGFR2 phosphorylation. [1] In hypoxia (5% O2), which upregulates VE‑PTP and blunts ANG1‑induced TIE2 activation, Razuprotafib restores TIE2 phosphorylation and downstream signaling even in the presence of exogenous ANG2. [1] In a permeability assay, 10 mM Razuprotafib blocks VEGF‑induced (200 ng/mL) FITC‑dextran (250 kDa) leakage across confluent HUVEC monolayers. [1] |
| ln Vivo |
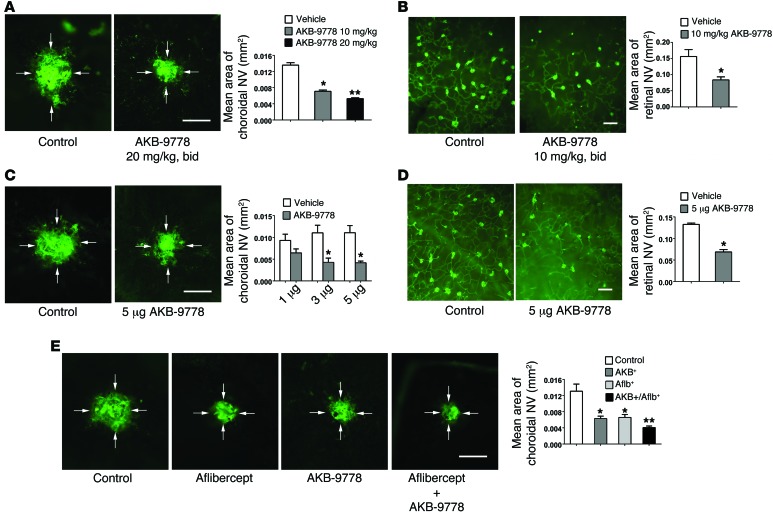
Subcutaneous injection of razuprotafib (20 mg/kg) stimulates TIE2 phosphorylation in retinal endothelial cells in vivo [1]. Subretinal neovascularization (NV) is inhibited by razuprotafib (10–20 mg/kg; subcutaneous injection; twice daily for 7 days) [1].
In the oxygen‑induced ischemic retinopathy mouse model, subcutaneous Razuprotafib (AKB-9778) at 10 or 20 mg/kg twice daily or intraocular injection (3–5 μg) significantly reduces retinal NV area. [1] In the laser‑induced choroidal NV model, subcutaneous Razuprotafib (10 or 20 mg/kg twice daily) or intraocular injection (3 or 5 μg) decreases choroidal NV area. Combination with intraocular aflibercept (40 μg) produces greater suppression than either agent alone. [1] In Rho‑VEGF transgenic mice (subretinal NV model), subcutaneous Razuprotafib (10 mg/kg twice daily) or intraocular injection (5 μg) reduces subretinal NV area. [1] In Tet‑opsin‑Ang2 mice with high ANG2 expression, intraocular Razuprotafib (5 μg) suppresses ischemia‑induced retinal NV despite high ANG2 levels. [1] In the Miles assay, intravenous Razuprotafib (16 mg/kg) significantly reduces histamine‑ and VEGF‑induced dermal vascular leakage. [1] In Rho‑VEGF mice, subcutaneous Razuprotafib (3 or 10 mg/kg) decreases albumin extravasation around retinal NV. [1] In Tet‑opsin‑VEGF double‑transgenic mice, subcutaneous Razuprotafib (10 or 50 mg/kg twice daily) prevents exudative retinal detachments and suppresses NV in a dose‑dependent manner. [1] |
| Enzyme Assay |
The phosphatase inhibition assay is performed in 96‑well plates. Test compounds are diluted in assay buffer (50 mM Tris‑HCl, 150 mM NaCl, 5 mM DTT, 1 mM EDTA, ±0.01% BSA, pH 7–10). Enzyme (commercially available or prepared proteins) is diluted in assay buffer immediately before use. Compound and enzyme are pre‑incubated for 10 minutes at room temperature. Then the fluorogenic phosphatase substrate DiFMUP (6,8‑difluoro‑4‑methylumbelliferyl phosphate) is added, followed by centrifugation at 500 g and incubation for 15 minutes at room temperature. The reaction is stopped by adding 5 mL of stop reagent (50 mM bpV[phen]). Plates are read on a plate reader. IC50 values are calculated using Excel Fit from concentration‑response curves. [1]
|
| Cell Assay |
Human umbilical vein endothelial cells (HUVECs, passages ≤6) are treated with Razuprotafib (AKB-9778) alone or with recombinant human ANG1 (500 ng/mL), ANG2 (500 ng/mL), or VEGF (200 ng/mL) for 10 minutes. For immunoprecipitation, cell lysates are incubated with anti‑TIE2 or anti‑VEGFR2 antibodies, then blotted with anti‑phosphotyrosine (4G10) or corresponding total protein antibodies. For signaling studies, lysates are probed with antibodies against total and phosphorylated AKT, ERK, and eNOS. For permeability assays, HUVECs are grown to confluence on Transwell filters, pretreated with 10 mM Razuprotafib or vehicle for 30 minutes, then 200 ng/mL VEGF and 250 kDa FITC‑dextran (0.25 mg/mL) are added to the upper chamber. After 2.5 hours, FITC‑dextran efflux into the lower chamber is measured using a plate reader. TIE2 silencing is performed using a retroviral shRNA specific for human TIE2 (sequence: 5'‑GATCCCACCATCGAGG‑3') in EC‑RF24 cells. [1]
|
| Animal Protocol |
Oxygen‑induced ischemic retinopathy: C57BL/6 mice at postnatal day 12 (P12) receive an intravitreous injection (1–2 μL) of Razuprotafib (AKB-9778) (0.1–5 μg) or vehicle. Alternatively, subcutaneous injections (10 or 20 mg/kg) are given twice daily from P12 to P17. At P17, eyes are fixed and retinas stained with FITC‑labeled GSA lectin, flat‑mounted, and NV area measured. [1]
Laser‑induced choroidal NV: Six‑week‑old C57BL/6 mice undergo Bruch’s membrane rupture by laser. Immediately after, mice receive intraocular injection of Razuprotafib (3 or 5 μg) or vehicle, or subcutaneous injections (10 or 20 mg/kg) twice daily for 7 days. In combination studies, intraocular aflibercept (40 μg) is given with subcutaneous Razuprotafib (20 mg/kg twice daily). After 7 days, choroidal flat mounts are stained with FITC‑GSA and NV area measured. [1] Rho‑VEGF transgenic mice (subretinal NV): At P15, mice receive subcutaneous Razuprotafib (3 or 10 mg/kg) twice daily until P21, or a single intraocular injection (5 μg). At P21, retinas are flat‑mounted and subretinal NV area quantified. For albumin leakage, mice receive three subcutaneous injections of Razuprotafib (3 or 10 mg/kg) 12 hours apart, then retinas stained for albumin and GSA. [1] Tet‑opsin‑Ang2 mice: Dox‑treated mice with ischemic retinopathy receive a single intraocular injection of Razuprotafib (5 μg) at P12. At P17, retinal NV area is measured. [1] Tet‑opsin‑VEGF double‑transgenic mice: Mice are pretreated with subcutaneous Razuprotafib (3, 10, or 50 mg/kg) twice daily for 3 days, then continued with the same regimen plus daily subcutaneous doxycycline (50 mg/kg) for 4 days. Eyes are sectioned and the percentage of retinal detachment measured. [1] Miles assay (vascular leakage in skin): C57BL/6 mice receive intravenous injection of Razuprotafib (16 mg/kg in 5% dextrose‑H₂O) or vehicle 5 hours before and immediately before the assay. Then Evans blue dye (1% in PBS, 100 μL) is injected intravenously. Ten minutes later, intradermal injections (50 μL) of PBS, histamine (225 ng), or mouse VEGF165 (100 ng) are given at three back sites. After 30 minutes, skin punches are excised and extracted in formamide for 5 days, and Evans blue extravasation is quantified. [1] |
| References | |
| Additional Infomation |
Razuprotafib, also known as AKB-9778, is a small molecule inhibitor that restores Tie2 activation by inhibiting VE-PTP. In studies of diabetes and COVID-19, Razuprotafib was self-administered by patients via subcutaneous injection. Razuprotafib is a small molecule inhibitor of vascular endothelial protein tyrosine phosphatase (VE-PTP) with potential vascular stabilizing effects. After administration, Razuprotafib targets, binds to, and inhibits VE-PTP, a negative regulator of endothelial cell (EC)-specific receptor tyrosine kinase (RTK) Tie2. This restores Tie2 activation, thereby improving endothelial function and stabilizing blood vessels. VE-PTP is upregulated in damaged endothelium associated with various diseases. Tie2 plays a crucial role in endothelial function and vascular stability. Reduced Tie2 activation leads to vascular leakage and inflammation.
Mechanism of Action Razuprotafib inhibits VE-PTP (a negative regulator of Tie2 in diseased blood vessels) by binding to and inhibiting its intracellular catalytic domain, thereby inactivating Tie2. This allows Razuprotafib to restore Tie2 activation, thereby enhancing endothelial function and stabilizing blood vessels. Razuprotafib is currently being investigated for its efficacy in treating diabetic vascular complications and COVID-19 acute respiratory distress syndrome (ARDS). Mechanism of Action Razuprotafib (AKB-9778) is a competitive inhibitor of VE‑PTP’s catalytic activity. It activates TIE2 even in the presence of high ANG2 levels, shifting ANG2 from antagonist to agonist. [1] Intraocular or systemic administration suppresses both choroidal and retinal NV, as well as vascular leakage, in multiple preclinical models. [1] Combination with aflibercept (VEGF trap) shows additive/synergistic effects in reducing choroidal NV. [1] Razuprotafib does not affect normal retinal vascular development (P4‑P7), suggesting selectivity for pathologic angiogenesis. [1] It has potential for treating neovascular AMD, diabetic retinopathy, diabetic macular edema, and retinal vein occlusion, especially in patients with incomplete response to anti‑VEGF therapy. [1] |
| Molecular Formula |
C26H26N4O6S3
|
|---|---|
| Molecular Weight |
586.702842235565
|
| Exact Mass |
586.101
|
| Elemental Analysis |
C, 53.23; H, 4.47; N, 9.55; O, 16.36; S, 16.39
|
| CAS # |
1008510-37-9
|
| Related CAS # |
1809275-69-1 (sodium);1008510-37-9;
|
| PubChem CID |
46700782
|
| Appearance |
White to off-white solid powder
|
| Density |
1.5±0.1 g/cm3
|
| Index of Refraction |
1.667
|
| LogP |
4.02
|
| Hydrogen Bond Donor Count |
4
|
| Hydrogen Bond Acceptor Count |
10
|
| Rotatable Bond Count |
12
|
| Heavy Atom Count |
39
|
| Complexity |
906
|
| Defined Atom Stereocenter Count |
2
|
| SMILES |
COC(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CC2=CC=C(C=C2)NS(=O)(=O)O)C3=CSC(=N3)C4=CC=CS4
|
| InChi Key |
KWJDHELCGJFUHW-SFTDATJTSA-N
|
| InChi Code |
InChI=1S/C26H26N4O6S3/c1-36-26(32)29-21(15-17-6-3-2-4-7-17)24(31)27-20(22-16-38-25(28-22)23-8-5-13-37-23)14-18-9-11-19(12-10-18)30-39(33,34)35/h2-13,16,20-21,30H,14-15H2,1H3,(H,27,31)(H,29,32)(H,33,34,35)/t20-,21-/m0/s1
|
| Chemical Name |
N-(4-{(2S)-2-{(2S)-2-[(methoxycarbonyl)amino]-3-phenylpropanamido}-2-[2-(thiophen-2-yl)-1,3-thiazol-4-yl]ethyl}phenyl)sulfamic acid
|
| Synonyms |
AKB-9778; AKB 9778; Razuprotafib; 1008510-37-9; AKB-9,778; 0WAX4UT396; AKB9,778; AKB9778
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO: ~100 mg/mL (170.4 mM)
|
|---|---|
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples.
Injection Formulations
Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline)(e.g. IP/IV/IM/SC) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). View More
Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] Oral Formulations
Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). View More
Oral Formulation 3: Dissolved in PEG400 (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7044 mL | 8.5222 mL | 17.0445 mL | |
| 5 mM | 0.3409 mL | 1.7044 mL | 3.4089 mL | |
| 10 mM | 0.1704 mL | 0.8522 mL | 1.7044 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved