| Size | Price | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
































| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion
/Monkeys/ maintained a state of continuous intoxication which was sustained when the dose /of PCP/ was increased to 0.5 mg/kg. Substitution of saline after 58 days of exposure resulted in the appearance of numerous abstinence signs and symptoms including increased vocalizations, bruxism, oculomotor hyperactivity, diarrhea, refusal of preferred food, piloerection, and tremors. Less common signs included ear and facial twitches, priapism, abdominal contractions, emesis, and convulsions. The time course of withdrawal was characterized by an initial recovery from the PCP-induced intoxication at about 4 hr after saline substitution. Onset of hyper-responsive behaviors became evident at 8-12 hr, with the maximum number of symptoms occurring 12-15 hr post-substitution. The syndrome dissipated over 24 hr, however, and all withdrawal signs were immediately reversed by PCP (0.25 g/kg, iv). This study also showed that PCP blood levels were in the 105-280 mg/mL range during self administration, and declined to 0-12 mg/mL with saline substitution. /MILK/ Excreted in milk. /From table/ /Phencyclidine/ is rapidly absorbed from the respiratory and the gastrointestinal tracts; as such, it is typically self-administered by oral ingestion, nasal insufflation, smoking, or IV and subcutaneous injection. Urine pH is an important determinant of renal elimination of PCP. In acidic urine, PCP becomes ionized and then cannot be reabsorbed. Acidification of the urine increased renal clearance of PCP from 1.98 + or- 0.48 L/hr to 2.4 + or - 0.78 L/hr. Additional studies have found a much higher renal clearance (8.04 + or- 1.56 L/hr) if the urine pH was decreased to less than 5.0. Although this may account for a 23% increase in the renal clearance, it only represents a 1.2% increase of the total clearance. For more Absorption, Distribution and Excretion (Complete) data for Phencyclidine (8 total), please visit the HSDB record page. Metabolism / Metabolites PCP is metabolized mainly in the liver. Oxidative hydroxylation to the inactive monohydroxypiperidine is followed by glucuronidation to a more water-soluble, conjugated derivative that is then excreted in the urine as the major form of metabolism. Significant first-pass liver metabolism also occurs when the drug is ingested orally, as opposed to being smoked or injected. Approximately 10% of drug is excreted unchanged un the urine. Abuse Liability: A bioassay procedure ... demonstrated that eight structurally related analogs of PCP were self administered by the dog. The thienyl-substituted analog was the most potent, followed by PCP, N-substituted alkyl analogs, monohydroxylated metabolites, and ketamine. Phencyclidine has known human metabolites that include PCHP and c-PPC/t-PPC. Biological Half-Life PCP elimination from plasma is consistent with first-order kinetics with a half life of 7 to 26 hours. The terminal phase serum half-life of PCP is 21 to 24 hours. /PCP/ has an apparent terminal half life of 21 + or - 3 hours under both control and overdose settings. Harmonic mean elimination half-lives of 14 hr (iv administration), 24 hr (oral administration), and 17 hr (administration via smoking) have been reported ... |
|---|---|
| Toxicity/Toxicokinetics |
Hepatotoxicity
Phencyclidine is no longer used medically and its production is outlawed. Nevertheless, phencyclidine remains an agent of abuse, used for its hallucinogenic effects. At low doses, phencyclidine appears to have little effect on the liver. However, high doses of phencyclidine have been associated with malignant hyperthermia which can trigger acute hepatitis necrosis and liver failure. Patients generally present with seizures and coma followed by severe hyperthermia, rhabdomyolysis and renal failure. The liver injury arises 1 to 2 days after the overdose with marked, rapid elevations in serum ALT, AST and LDH, with minimal increases in alkaline phosphatase and delayed rises in bilirubin (Case 1). Coma arises early along with prolongation of the prothrombin time, hyperammonemia and metabolic acidosis. The abnormalities resolve almost as rapidly as they develop and with suitable life support, survival is not uncommon (Case 1). The clinical syndrome is that of acute hepatic necrosis and resembles the acute liver injury that occurs with heat shock, severe hypoxia and hepatic ischemia. Liver biopsy shows severe centrilobular necrosis with mild inflammation. Likelihood score: C[HD] (probable cause of clinically apparent acute liver injury but only when given in high doses). Mechanism of Injury The mechanism of acute liver injury by phencyclidine is probably hyperthermia, hypoxia and hypotension, and the clinical course and outcome resembles that associated with shock or severe hypoxia. Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation A single case of phencyclidine use has been reported in which a small amount of phencyclidine was detected in breastmilk over 6 weeks after use of an unknown quantity of the drug. Effects on the breastfed infant are unknown. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. |
| Additional Infomation |
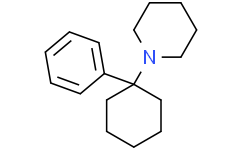
Phencyclidine is a member of the class of piperidines that is piperidine in which the nitrogen is substituted with a 1-phenylcyclohexyl group. Formerly used as an anaesthetic agent, it exhibits both hallucinogenic and neurotoxic effects. It has a role as a neurotoxin, a psychotropic drug, an anaesthetic and a NMDA receptor antagonist. It is a member of piperidines and a member of benzenes. It derives from a hydride of a piperidine.
Phencyclidine is a DEA Schedule II controlled substance. Substances in the DEA Schedule II have a high potential for abuse which may lead to severe psychological or physical dependence. It is a Depressants substance. A hallucinogen formerly used as a veterinary anesthetic, and briefly as a general anesthetic for humans. Phencyclidine is similar to ketamine in structure and in many of its effects. Like ketamine, it can produce a dissociative state. It exerts its pharmacological action through inhibition of NMDA receptors (receptors, N-methyl-D-aspartate). As a drug of abuse, it is known as PCP and Angel Dust. Phencyclidine is an illegal, hallucinogenic drug that was initially used as an anesthetic agent in the 1950s and early 1960s, but was then withdrawn in 1965 because of dissociative hallucinogenic effects that were often disturbing and sometimes severe and prolonged. The “out-of-body” intense psychological and behavioral effects of low doses of phencyclidine led to its abuse. In the late 1960s and 1970s, phencyclidine (“angel dust”) became a widely used hallucinogenic drug. The effects were often extreme, marked by acute psychosis and aggressive and violent behaviors, and overdoses led to many emergency room visits and deaths from status epilepticus, hyperthermia, rhabdomyolysis and subsequent renal, respiratory and hepatic failure. Phencyclidine is developed as an anesthetic for humans in 1959. Its use was discontinued due to extreme side effects that included delirium, confusion, visual disturbances, hallucinations and violence; some evidence of long-term memory disorders and schizophrenia-like syndrome has been observed. Phencyclidine, a substance of abuse also know as 'angel dust', can cause physical and psychological distresses, such as coma, seizures, convulsions, respiratory depression, and cardiac problems. A hallucinogen formerly used as a veterinary anesthetic, and briefly as a general anesthetic for humans. Phencyclidine is similar to KETAMINE in structure and in many of its effects. Like ketamine, it can produce a dissociative state. It exerts its pharmacological action through inhibition of NMDA receptors (RECEPTORS, N-METHYL-D-ASPARTATE). As a drug of abuse, it is known as PCP and Angel Dust. Mechanism of Action The N-methyl-D-Aspartate (NMDA) receptor, a type of ionotropic receptor, is found on the dendrites of neurons and receives signals in the form of neurotransmitters. It is a major excitatory receptor in the brain. Normal physiological function requires that the activated receptor fluxes positive ions through the channel part of the receptor. PCP enters the ion channel from the outside of the neuron and binds, reversibly, to a site in the channel pore, blocking the flux of positive ions into the cell. PCP therefore inhibits depolarization of neurons and interferes with cognitive and other functions of the nervous system. Noncompetitive N-methyl-d-aspartate (NMDA) receptor antagonists evoke a behavioral and neurobiological syndrome in experimental animals. We previously reported that phencyclidine (PCP), an NMDA receptor antagonist, increased locomotor activity in wildtype (WT) mice but not GluN2D subunit knockout mice. Thus, the aim of the present study was to determine whether the GluN2D subunit is involved in PCP-induced motor impairment. PCP or UBP141 (a GluN2D antagonist) induced potent motor impairment in WT mice but not GluN2D KO mice. By contrast, CIQ, a GluN2C/2D potentiator, induced severe motor impairment in GluN2D KO mice but not WT mice, suggesting that the GluN2D subunit plays an essential role in the effects of PCP and UBP141, and an appropriate balance between GluN2C and GluN2D subunits might be needed for appropriate motor performance. The level of the GluN2D subunit in the mature mouse brain is very low and restricted. GluN2D subunits exist in brainstem structures, the globus pallidus, thalamus, and subthalamic nucleus. We found that the expression of the c-fos gene increased the most among PCP-dependent differentially expressed genes between WT and GluN2D KO mice, and the number of Fos-positive cells increased after PCP administration in the basal ganglia motor circuit in WT mice but not GluN2D KO mice. These results suggest that the GluN2D subunit within the motor circuitry is a key subunit for PCP-induced motor impairment, which requires an intricate balance between GluN2C- and GluN2D-mediated excitatory outputs. Prepulse inhibition (PPI) is an example of sensorimotor gating and deficits in PPI have been demonstrated in schizophrenia patients. Phencyclidine (PCP) suppression of PPI in animals has been studied to elucidate the pathological elements of schizophrenia. However, the molecular mechanisms underlying PCP treatment or PPI in the brain are still poorly understood. In this study, quantitative phosphoproteomic analysis was performed on the prefrontal cortex from rats that were subjected to PPI after being systemically injected with PCP or saline. PCP downregulated phosphorylation events were significantly enriched in proteins associated with long-term potentiation (LTP). Importantly, this data set identifies functionally novel phosphorylation sites on known LTP-associated signaling molecules. In addition, mutagenesis of a significantly altered phosphorylation site on xCT (SLC7A11), the light chain of system xc-, the cystine/glutamate antiporter, suggests that PCP also regulates the activity of this protein. Finally, new insights were also derived on PPI signaling independent of PCP treatment. This is the first quantitative phosphorylation proteomic analysis providing new molecular insights into sensorimotor gating. Phencyclidine (PCP), a noncompetitive N-methyl-D-aspartate receptor antagonist, induces psychotomimetic effects in humans and animals. Administration of PCP to rodents is used as a preclinical model for schizophrenia; however, the molecular mechanisms underlying the symptoms remain largely unknown. Acute PCP treatment rapidly induces behavioral and cognitive deficits; therefore, post-translational regulation of protein activity is expected to play a role at early time points. We performed mass-spectrometry-driven quantitative analysis of rat frontal cortex 15, 30, or 240 min after the administration of PCP (10 mg/kg). We identified and quantified 23,548 peptides, including 4749 phosphopeptides, corresponding to 2604 proteins. A total of 352 proteins exhibited altered phosphorylation levels, indicating that protein phosphorylation is involved in the acute response to PCP. Computational assessment of the regulated proteins biological function revealed that PCP perturbs key processes in the frontal cortex including calcium homeostasis, organization of cytoskeleton, endo/exocytosis, and energy metabolism. This study on acute PCP treatment provides the largest proteomics and phosphoproteomics data sets to date of a preclinical model of schizophrenia. Our findings contribute to the understanding of alterations in glutamatergic neurotransmission in schizophrenia and provide a foundation for discovery of novel targets for pharmacological intervention. Phencyclidine (PCP) is a psychotomimetic drug that induces schizophrenia-like symptoms in healthy individuals and exacerbates pre-existing symptoms in patients with schizophrenia. PCP also induces behavioral and cognitive abnormalities in non-human animals, and PCP-treated animals are considered a reliable pharmacological model of schizophrenia. However, the exact neural mechanisms by which PCP modulates behavior are not known. During the last decade several studies have indicated that disturbed activity of the prefrontal cortex (PFC) may be closely related to PCP-induced psychosis. Systemic administration of PCP produces long-lasting activation of medial PFC (mPFC) neurons in rats, almost in parallel with augmentation of locomotor activity and behavioral stereotypies. Later studies have showed that such PCP-induced behavioral abnormalities are ameliorated by prior administration of drugs that normalize or inhibit excess excitability of PFC neurons. Similar activation of mPFC neurons is not induced by systemic injection of a typical psychostimulant such as methamphetamine, even though behavioral hyperactivity is induced to almost the same level. This suggests that the neural circuits mediating PCP-induced psychosis are different to those mediating methamphetamine-induced psychosis. Locally applied PCP does not induce excitation of mPFC neurons, indicating that PCP-induced tonic excitation of mPFC neurons is mediated by inputs from regions outside the mPFC. This hypothesis is strongly supported by experimental results showing that local perfusion of PCP in the ventral hippocampus, which has dense fiber projections to the mPFC, induces tonic activation of mPFC neurons with accompanying augmentation of behavioral abnormalities. In this review we summarize current knowledge on the neural mechanisms underlying PCP-induced psychosis and highlight a possible involvement of the PFC and the hippocampus in PCP-induced psychosis. For more Mechanism of Action (Complete) data for Phencyclidine (11 total), please visit the HSDB record page. |
| Molecular Formula |
C17H25N
|
|---|---|
| Molecular Weight |
243.3871
|
| Exact Mass |
243.198
|
| CAS # |
77-10-1
|
| Related CAS # |
2981-31-9 (hydrobromide);77-10-1 (hydrochloride);77-10-1 (Parent)
|
| PubChem CID |
6468
|
| Appearance |
White, crystalline powder
On the street may contain a number of contaminants causing the color to range from tan to brown with a consistency ranging from powder to a gummy mass Colorless crystals White crystalline powder |
| LogP |
3.6
|
| Hydrogen Bond Donor Count |
0
|
| Hydrogen Bond Acceptor Count |
1
|
| Rotatable Bond Count |
2
|
| Heavy Atom Count |
18
|
| Complexity |
242
|
| Defined Atom Stereocenter Count |
0
|
| InChi Key |
JTJMJGYZQZDUJJ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C17H25N/c1-4-10-16(11-5-1)17(12-6-2-7-13-17)18-14-8-3-9-15-18/h1,4-5,10-11H,2-3,6-9,12-15H2
|
| Chemical Name |
1-(1-phenylcyclohexyl)piperidine
|
| Synonyms |
Angel mist; Angel dust; Phencyclidine
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples.
Injection Formulations
Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline)(e.g. IP/IV/IM/SC) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). View More
Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] Oral Formulations
Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). View More
Oral Formulation 3: Dissolved in PEG400 (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.1086 mL | 20.5432 mL | 41.0863 mL | |
| 5 mM | 0.8217 mL | 4.1086 mL | 8.2173 mL | |
| 10 mM | 0.4109 mL | 2.0543 mL | 4.1086 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved


























