| Size | Price | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
































| Targets |
P. falciparum NF54 (IC50 = 543 nM)
|
|---|---|
| ln Vitro |
In Vitro Antiplasmodial Activity [1]
All synthesized compounds were tested for in vitro growth inhibitory activity against a drug-sensitive strain of P. falciparum, NF54, and selected compounds were tested for activity against a multidrug resistant strain, K1, with chloroquine and artesunate as positive controls. Compounds with good antiplasmodial activity (IC50s < 200 nM) were also evaluated for in vitro mammalian cytotoxicity in the Chinese Hamster Ovary (CHO) cell line, using emetine as a positive control. The preliminary structure–activity relationship (SAR) investigation was focused on identifying substituents at the 2- and 8-positions of the 1,5-naphthyridine ring that gave potent antiplasmodial activity (Table 1). At R1, the general SAR trend showed that pyridine analogues (15–19) displayed improved activity compared to other heterocyclic substituents like pyrazole (8, 9), phenyl (10–14), and piperazine (20–22). Both 3- and 4-linked pyridines (16, 17) were equally potent and small substitutions on the pyridine rings (18, 19) were tolerated. For optimization of antiplasmodial activity, by making changes at the 2-position of the 1,5-naphthyridine ring, the analogue with the 4-pyridyl group at position 8 of the scaffold was synthesized first and maintained for matched pair analyses (Table 2). This work was facilitated by the synthetic accessibility of multigram quantities of the required pyridine precursor, intermediate 5. As can be seen in Tables 1 and 2, replacing the pyridine sulfonamide at the 2-position of the naphthyridine ring (R2), e.g., compound 8 (NF54 IC50 = 543 nM) and compound 15 (NF54 IC50 = 377 nM), with phenyl sulfonamide in the meta-position as in compounds 9 (NF54 IC50 = 277 nM) and 16 (NF54 IC50 = 122 nM), improved the NF54 activity 2- to 3-fold. ortho- and para-Phenyl sulfonamides diminished NF54 activity for compounds 23, 24, and 31 (IC50s > 5000 nM). The carboxamide, as in compound 37, was not as well-tolerated (NF54 IC50 = 745 nM) compared to the sulfonamide matched pair, compound 16. Overall, aryl carboxamides 37–41 and amines 27, 43–45 at R2 displayed diminished antiplasmodial activity. |
| ln Vivo |
Compound 55 [ an analog of MMV024101] was assessed for in vivo efficacy using the NSG mouse model of P. falciparum infection (Pf3D7 IC50 = 142 nM). NSG mice are genetically immunodeficient and thereby able to support engraftment by human red blood cells and infection by the human specific P. falciparum parasite. (24) A quadruple-dose regimen of 50 mg·kg–1 of 55 for four consecutive days showed 80% reduction in parasitemia compared to untreated mice (Figure 3). The pharmacokinetics of 55 from the study showed good exposure and a dose-dependent correlation with the reduction in parasitemia (Table 8 and Figure 4). However, 55 had relatively slow killing kinetics (see below) and was cleared quickly. This likely accounts for parasitemia remaining flat throughout the course of the experiment.[1]
Pharmacokinetic Studies [1] When dosed intravenously, 30/ an analog of MMV024101 was cleared quickly from blood (90 mL·min–1·kg–1). Tissue distribution was high (25.7 L·kg–1), and half-life was moderate (3.3 h) (Table 7). A comparative oral pharmacokinetic study of 30 between mice pretreated with 1-aminobenzotriazole (ABT, a nonselective CYP inhibitor and untreated mice was carried out to ascertain whether or not the oral exposure of the compound could be improved, and metabolism of 30 into the less active metabolite 16 minimized. Without ABT, 30 was absorbed quickly (Tmax of 0.5 h), but the oral exposure was low with an oral bioavailability of 8%. Exposure of the metabolite 16 was 10-fold the exposure of the parent 30, but with a short half-life (1.6 h). ABT did partially inhibit the biotransformation of 30 into 16, with a two-fold increase and two-fold decrease of the oral exposure of the parent and metabolite, respectively. As a result, the oral bioavailability of 30 did not improve significantly (15%). In fact, ABT is known to inhibit CYP enzymatic activity but not completely. (23) These results suggested that metabolism was not the only limiting factor with respect to oral exposure of 30 and that other clearance mechanisms were involved. As 16 was about 6-fold less active than 30, it was not expected to contribute significantly to the pharmacodynamic profile of 30, and the latter was not progressed to efficacy studies. |
| Enzyme Assay |
Human protein kinase screening data and experimental [1]
The principal method utilized is a radioactive filter binding assay using 33P ATP. This method is sensitive, accurate and provides a direct measure of activity. Compounds were added to a ‘mother plate’ that serves as the source for ‘daughter plates’ which will be stored at -20 ⁰C until assay initiation. All compounds were screened in duplicate. There will be 3 additions to the assay; First, Enzyme-substrate mixture followed by an incubation time of 5 minutes at room temperature. Second, 33P ATP was added which begins the assay followed by a varied incubation period (based on enzyme) at room temperature. Finally, orthophosphoric acid addition will halt the assay. Assay components were harvested onto P81 filter plates and later were air-dried. Scintillation fluid was added to plates and counts were S23 read on a Topcount NXT. Finally, a mean percentage activity is calculated for each the assayed compounds. Test samples were screened for in vitro antiplasmodial activity against a chloroquine sensitive (CQS) strain (NF54) and multidrug-resistant (K1) strain of the malaria parasite P. falciparum. Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified version of the method of Trager and Jensen (1976).1 Quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method described by Makler (1993).2 The test samples were tested in triplicate on two separate occasions. Further dilutions were prepared in complete medium on the day of the experiment. Samples were tested as a suspension if not completely dissolved. Chloroquine and artesunate were used as the reference drugs [1]. The metabolic stability assay was performed in duplicate in a 96-well microtiter plate. The test compounds (0.1 µM) were incubated (37 °C) in mouse, rat and pooled human liver microsomes (final protein concentration of 0.4 mg/mL; XenoTech, Lenexa, KS) suspended in 0.1 M phosphate buffer (pH 7.4) for predetermined time points, in the presence and absence of the cofactor NADPH (1 mM). The reactions were quenched by the addition of ice cold S19 acetonitrile containing internal standard (carbamazepine, 0.0236 µg/mL). The samples were centrifuged and the supernatant was filtered and analyzed by means of LC-MS/MS (Agilent Rapid Resolution HPLC, AB SCIEX 4500 MS). The relative loss of parent compound over time was monitored and plots were prepared for each compound of concentration versus time to determine the first order rate constant for compound depletion [1]. Plasma protein binding assay. [1] The plasma protein binding assay was performed in a 96-well microtiter plate with pooled human plasma, spiked with test compound (5 µM). An aliquot was immediately removed and quenched using ice cold acetonitrile containing internal standard (carbamazepine, 0.0236 µg/mL), and placed in the freezer. This served as the total concentration sample. After preincubation (37 °C for 1 hour) duplicate aliquots of the spiked plasma were transferred to ultra-centrifugation tubes, and ultracentrifuged for 4 hours (42000 rpm, 37 °C). Thus obtained supernatant was collected and analyte concentration of all compounds and sample types were determined by means of LC-MS/MS. |
| Cell Assay |
In vitro cytotoxicity assay experimental. [1]
Compounds were tested for in vitro cytotoxicity against a mammalian cell-line, Chinese Hamster Ovarian (CHO) using the 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazoliumbromide (MTT)-assay. The MTT-assay was used as a colorimetric assay for cellular growth and survival, and compares well with other available assays. The tetrazolium salt MTT was used to measure all growth and chemosensitivity. The test samples were tested in triplicate on one occasion. The test samples were prepared to a 10 mM stock solution in DMSO and were tested as a suspension if not properly dissolved. Emetine was used as the reference drug in all experiments. The initial concentration of emetine was 100 µg/mL, which was serially diluted in complete medium with 10-fold dilutions to give 6 concentrations, the lowest being 0.001 µg/mL. The same dilution technique was applied to the all test samples. The highest concentration of solvent to which the cells were exposed to have no measurable effect on the cell viability. The 50% inhibitory concentration (IC50) values were obtained from full dose-response curves, using a non-linear dose-response curve fitting analysis via GraphPad Prism v.4 software. Permeability across Caco-2 cell monolayers. [1] Caco-2 cells were seeded onto 0.3 cm2 polycarbonate filter transwells at a density of 60,000 cells per well. The transport experiment was conducted using confluent cell monolayers on days 22-23 post-seeding. On the day of the permeability study, the integrity of the cell monolayers was determined by measuring the transepithelial electrical resistance (TEER) in the presence of pH 7.4 Hanks balanced salt solution, and only monolayers with TEER values of >400 Ω.cm2 were selected for use in the study. Permeability experiments were performed using human plasma as the transport medium in both the apical and basolateral chambers to overcome issues associated with poor mass balance of (-)-1 when using buffer as the transport medium. The permeability of marker compounds (i.e. 14C-mannitol, 3H-propranolol, and 3H-digoxin) was also assessed using the plasma transport medium with a subset of wells from the same batch to ensure assay performance. Donor solutions were prepared by spiking stock solutions of marker compounds into human plasma (final DMSO concentration of 0.1% v/v) at a nominal concentration of 20 µM. The solution was equilibrated at 37 °C for 4 hours before centrifuging for 5 minutes to remove any compound that may have precipitated. The visually clear supernatant plasma was then added to the transport wells. Samples from the donor chamber were taken at the start (within 2 minutes) and end of the experiment. Compound flux was determined over a period of 240 minutes with samples taken from the acceptor chamber at 6-9 time points. At each sample time, the volume of acceptor plasma removed was replaced with blank plasma. Acceptor concentrations were then corrected for the dilution that occurred with plasma replacement. Donor and acceptor samples were stored frozen at -80 °C until analysis by LC-MS. The measured initial donor concentration (CD) was corrected for the free concentration using the measured fraction unbound (fu) and the apparent permeability coefficient (Papp) was calculated using the following equation: Papp = dQ/dt × [1 / (CD×fu) × A] where dQ/dt is the apparent steady-state flux and A is the monolayer surface area. The efflux ratio was calculated as the ratio B-A Papp/A-B Papp. Gametocytocidal activity assay [1] Compound inhibitory activity was determined by preparing test samples in parasite culture medium in transparent 96-well flat bottom plates at 5 µM concentration (n = 2 for each data point). Parasitized red blood cells (Late and Early stage gametocytes) were added to a final concentration of 5% haematocrit, 2% gametocytemia and the plates were incubated for 48 hours before proceeding with the pLDH assay. Percentage parasite/gametocyte survival in each well was calculated relative to control wells that received no drug. |
| Animal Protocol |
Prior to animal studies being carried out, the research protocol was evaluated and approved by the Animal Ethics Committee at the University of Cape Town (protocols #013/028 and 011/049). The in vivo pharmacokinetic properties of 30 and 55 were studied following an orally administered (po) dose of 20 mg/kg and intravenous (iv) dose of 2 mg/kg for 30 and 5 mg/kg (po) and 1.5 mg/kg (iv) for 55. Compounds 30 and 55 were dissolved in DMA/PPG/PEG: 10/60/30 for iv dose and in 0.5% (w/v) hydroxypropylmethylcellulose (HPMC) water solution for oral dose, administered to male BALB/c mice (n=3 for each group). 1-aminobenzotriazole (ABT) dissolved in 0.5% (w/v) hydroxypropylmethylcellulose (HPMC) water solution was also co-administered orally at 50 mg/kg to mice 1 hour before the oral dose of 30. Blood samples were collected at predetermined sampling times, 0.17 h, 0.5 h, 1 h, 3 h, 5 h, 7 h, 10 h and 24 h for intravenous dosing; 0.5 h, 1 h, 3 h, 5 h, 7 h, 10 h, 24 h for oral dosing via tail bleeding in heparinized tubes and were stored at -80 °C until extraction. [1]
|
| ADME/Pharmacokinetics |
Pharmacokinetic Studies [1]
When dosed intravenously, 30/ an analog of MMV024101 was cleared quickly from blood (90 mL·min–1·kg–1). Tissue distribution was high (25.7 L·kg–1), and half-life was moderate (3.3 h) (Table 7). A comparative oral pharmacokinetic study of 30 between mice pretreated with 1-aminobenzotriazole (ABT, a nonselective CYP inhibitor and untreated mice was carried out to ascertain whether or not the oral exposure of the compound could be improved, and metabolism of 30 into the less active metabolite 16 minimized. Without ABT, 30 was absorbed quickly (Tmax of 0.5 h), but the oral exposure was low with an oral bioavailability of 8%. Exposure of the metabolite 16 was 10-fold the exposure of the parent 30, but with a short half-life (1.6 h). ABT did partially inhibit the biotransformation of 30 into 16, with a two-fold increase and two-fold decrease of the oral exposure of the parent and metabolite, respectively. As a result, the oral bioavailability of 30 did not improve significantly (15%). In fact, ABT is known to inhibit CYP enzymatic activity but not completely. (23) These results suggested that metabolism was not the only limiting factor with respect to oral exposure of 30 and that other clearance mechanisms were involved. As 16 was about 6-fold less active than 30, it was not expected to contribute significantly to the pharmacodynamic profile of 30, and the latter was not progressed to efficacy studies. |
| References |
[1]. Identification, Characterization, and Optimization of 2,8-Disubstituted-1,5-naphthyridines as Novel Plasmodium falciparum Phosphatidylinositol-4-kinase Inhibitors with in Vivo Efficacy in a Humanized Mouse Model of Malaria. J Med Chem. 2018 Jul 12;61(13):5692-5703.
|
| Additional Infomation |
A novel 2,8-disubstituted-1,5-naphthyridine hit compound stemming from the open access Medicines for Malaria Venture Pathogen Box formed a basis for a hit-to-lead medicinal chemistry program. Structure-activity relationship investigations resulted in compounds with potent antiplasmodial activity against both chloroquine sensitive (NF54) and multidrug resistant (K1) strains of the human malaria parasite Plasmodium falciparum. In the humanized P. falciparum mouse efficacy model, one of the frontrunner compounds showed in vivo efficacy at an oral dose of 4 × 50 mg·kg-1. In vitro mode-of-action studies revealed Plasmodium falciparum phosphatidylinositol-4-kinase as the target.
Starting from an open source hit compound, MMV024101 (compound 8) from the MMV Pathogen Box, a series of 2,8-disubstituted-1,5-naphthyridine analogues was synthesized and evaluated for in vitro antiplasmodial activity. From a total of 48 analogues made for SAR investigation, 26 showed improved blood stage activity compared to the original hit. Several analogues exhibited low nanomolar activity (NF54 IC50s < 100 nM) and were equipotent against both NF54 and K1. Two compounds, 21 and 26, were cross-resistant with a Pfpi4k laboratory mutant strain pointing to PI4K as the mode-of-action. The compounds were confirmed as potent inhibitors of PvPI4K using a biochemical assay. The realization that the compounds operate on PI4K is significant in that the importance of the target is magnified by MMV390048 now in Phase IIa clinical trials. Major routes of metabolism of the 2- and 8-substituents of the naphthyridine ring were blocked en route to delivering the lead compound 55. Compound 55 showed improved bioavailability in the mouse and was chosen for in vivo efficacy studies based on the pharmacokinetics and antiplasmodial activity. In the PfSCID mouse model of infection, 55 resulted in 80% reduction in parasitemia at 4 × 50 mg·kg–1. At this juncture, it is noteworthy that this compound meets the MMV Late Lead criteria with respect to CYP inhibition (IC50 > 20 μM), oral bioavailability (>30%), and hERG (>10 μM) but does not meet the in vitro antiplasmodial potency (IC50 < 10 nM) criteria. By contrast, MMV390048 shows much higher efficacy with a similar reduction in parasitemia at 4 × 1 mg·kg–1 and achieved 100% reduction in parasitemia at higher doses. This is a consequence of the much better pharmacokinetic profile and higher asexual blood stage activity of MMV390048. (10) Future optimization studies will aim to further improve pharmacokinetics and in vivo potency to enhance efficacy and to achieve the complete clearance of the malaria parasite to be comparable to the other PfPI4K inhibitors in development. [1] |
| Molecular Formula |
C16H12N6O2S
|
|---|---|
| Molecular Weight |
352.37
|
| Exact Mass |
352.074
|
| Elemental Analysis |
C, 54.54; H, 3.43; N, 23.85; O, 9.08; S, 9.10
|
| CAS # |
1092565-44-0
|
| PubChem CID |
25127876
|
| Appearance |
Typically exists as solid at room temperature
|
| LogP |
0.3
|
| Hydrogen Bond Donor Count |
2
|
| Hydrogen Bond Acceptor Count |
7
|
| Rotatable Bond Count |
3
|
| Heavy Atom Count |
25
|
| Complexity |
568
|
| Defined Atom Stereocenter Count |
0
|
| SMILES |
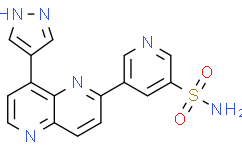
C1=CC2=NC=CC(=C2N=C1C3=CC(=CN=C3)S(=O)(=O)N)C4=CNN=C4
|
| InChi Key |
GTWOKQIGBJADIZ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C16H12N6O2S/c17-25(23,24)12-5-10(6-18-9-12)14-1-2-15-16(22-14)13(3-4-19-15)11-7-20-21-8-11/h1-9H,(H,20,21)(H2,17,23,24)
|
| Chemical Name |
5-[8-(1H-pyrazol-4-yl)-1,5-naphthyridin-2-yl]pyridine-3-sulfonamide
|
| Synonyms |
MMV 024101; MMV024101; 1092565-44-0; 5-[8-(1H-pyrazol-4-yl)-1,5-naphthyridin-2-yl]pyridine-3-sulfonamide; TCMDC-134293; 3-Pyridinesulfonamide, 5-[8-(1H-pyrazol-4-yl)-1,5-naphthyridin-2-yl]-; CHEMBL530228; SCHEMBL3111909; MMV-024101; MMV024101
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples.
Injection Formulations
Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline)(e.g. IP/IV/IM/SC) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). View More
Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] Oral Formulations
Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). View More
Oral Formulation 3: Dissolved in PEG400 (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8379 mL | 14.1896 mL | 28.3793 mL | |
| 5 mM | 0.5676 mL | 2.8379 mL | 5.6759 mL | |
| 10 mM | 0.2838 mL | 1.4190 mL | 2.8379 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved


























