| Size | Price | Stock | Qty |
|---|---|---|---|
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg | |||
| 500mg | |||
| Other Sizes |
































| Targets |
FEMIB E3 ligase; covalent binding mode targeting cysteine
|
|---|---|
| ln Vitro |
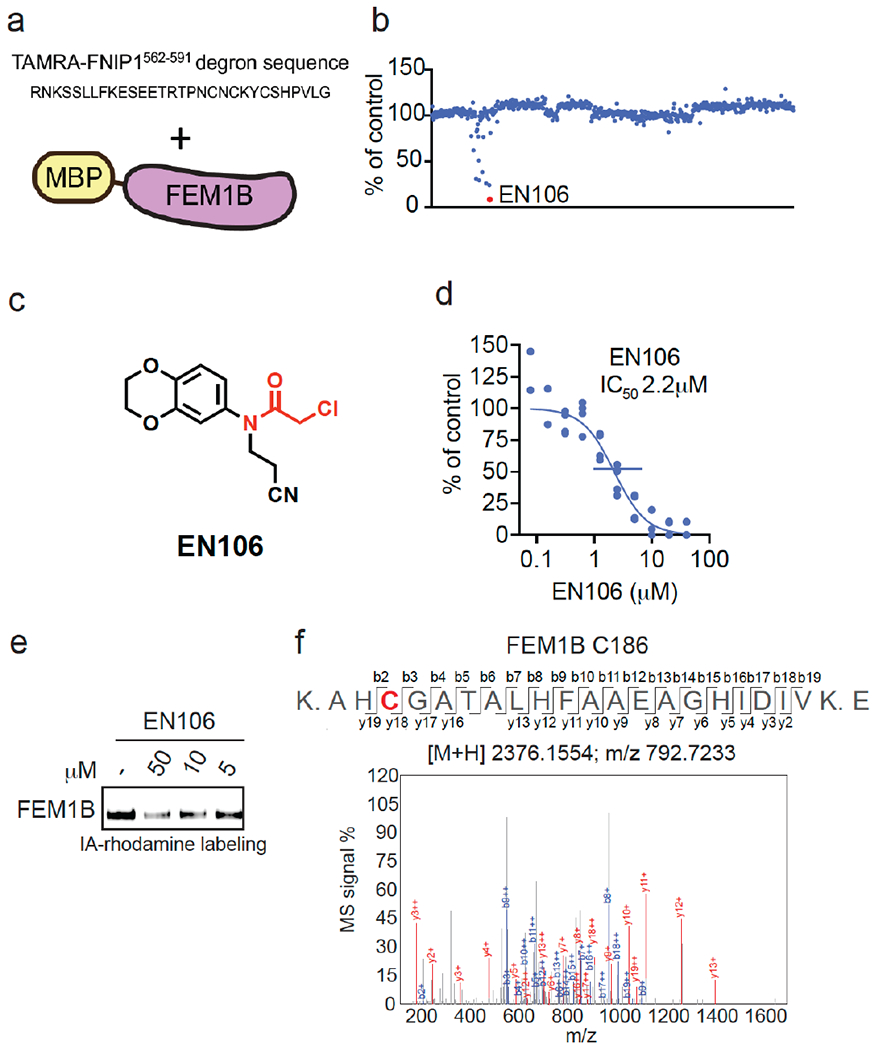
To identify a covalent FEM1B recruiter, we screened a library of 566 cysteine-reactive covalent ligands in a competitive fluorescence polarization assay using a TAMRA-conjugated FNIP1562-591 degron and recombinant mouse FEM1B (Figure 1a–1b, Table S1). Through this screen, we identified the chloroacetamide EN106 that inhibited FEM1B-FNIP1 degron fluorescence polarization with a 50 % inhibitory concentration (IC50) of 2.2 μM (Figure 1c–1d). From the initial screen, EN106 showed the most significant inhibition of FEM1B interactions with the FNIP1 degron (Figure 1b). EN106 showed competition against labeling of FEM1B with a cysteine-reactive rhodamine-conjugated iodoacetamide (IA-rhodamine) probe by gel-based ABPP, confirming a direct interaction of EN106 with a cysteine on FEM1B (Figure 1e). Analysis of EN106 reactivity with recombinant FEM1B by liquid-chromatography-tandem mass spectrometry (LC-MS/MS) analysis of FEM1B tryptic digests revealed an EN106 adduct only on C186—the site that was previously shown to be critical for FEM1B substrate recognition (Figure 1f). We also demonstrated that a non-reactive version of EN106, NJH-2-082, does not inhibit FEM1B interactions with the FNIP1 degron, confirming the importance of the covalent interactions of the cysteine-reactive warhead with C186 (Figure S1). [1]
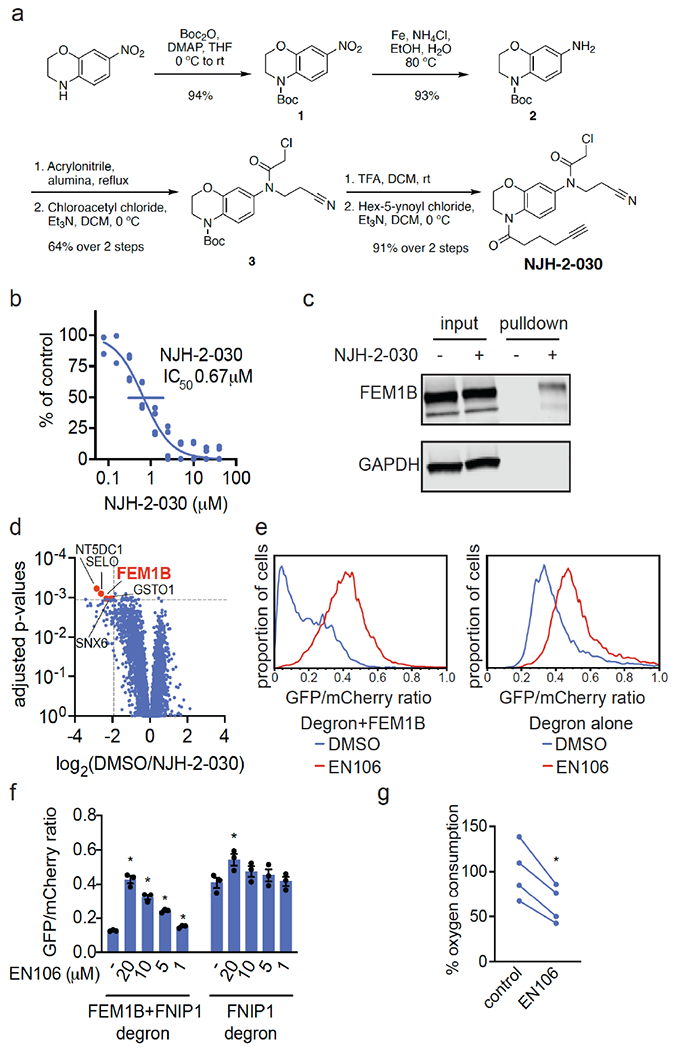
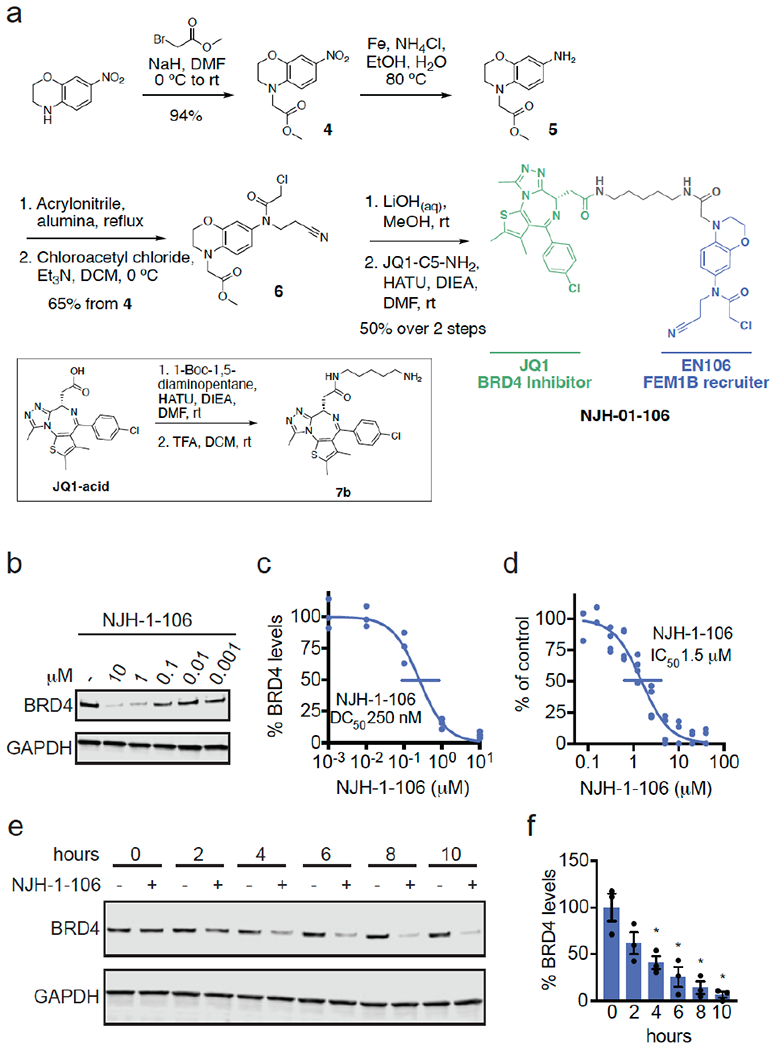
To confirm that EN106 engaged FEM1B in cells, we synthesized NJH-2-030, an alkyne-functionalized derivative of EN106 (Figure 2a). To maintain engagement of C186, the alkyne was positioned distal to the chloroacetamide by exchanging the benzodioxan for a dihydro[1,4]benzoxazine scaffold. The starting benzoxazine was Boc-protected to give 1 before reduction of the nitro group to provide aniline 2. Alkylation of 2 with acrylonitrile provided the propionitrile-substituted compound, which was acylated to obtain the chloroacetamide 3. Boc deprotection and acylation with hex-5-ynoyl chloride provided alkyne probe NJH-2-030 (Figure 2a). [1] To assess the proteome-wide cysteine-reactivity of EN106, we also performed a competitive isotopic tandem orthogonal proteolysis-ABPP (isoTOP-ABPP) study to quantitatively assess proteome-wide cysteine reactivity of EN106 in cells (Figure S2; Table S2). While we did not capture FEM1B in our chemoproteomics experiment, we only observed two targets of EN106 in cells—C63 of HNRNPA3 and C127 of PRDX3—across 1465 quantified cysteines (Figure S2). Neither of these off-targets of EN106 were E3 ligases. Given that we did not detect C186 of FEM1B in isoTOP-ABPP experiments, we also used our NJH-2-030 probe in situ in HEK293T cells to perform pulldown quantitative proteomic experiments to further confirm target engagement and proteome-wide selectivity of this close EN106 derivative (Figure 2d; Table S3). FEM1B was one of the most significantly enriched targets by the NJH-2-030 probe compared to DMSO controls with four additional off-targets detected—GSTO1, SNX6, SELO, and NT5DC1—of which none of these proteins were E3 ligases (Figure 2d; Table S3). These data collectively showed that EN106 or its derivatives functionally engaged FEM1B in cells without detectable off-target effects on other endogenous degradation pathway components. [1] To further demonstrate that EN106 disrupted substrate recognition by FEM1B in cells, we monitored the degradation of GFP linked to a FNIP1 degron compared to IRES driven expression of mCherry from the same plasmid in HEK293T cells by flow cytometry. EN106 treatment significantly stabilized FNIP1 degron-GFP levels, compared to vehicle-treated controls in a dose-responsive manner in FEM1B overexpressing cells (Figure 2e, f). EN106 increased FNIP1 reporter levels in cells lacking exogenously expressed FEM1B to a similar extent as previously observed upon deletion of FEM1B, indicating that this compound can target the endogenous E3 ligase (Figure 2e–2f). EN106 did not affect the pomalidomide-induced degradation of an unrelated E4F1 degron by the E3 ligase cereblon (Figure S3). These findings thus indicate that EN106 not only engages, but also inhibits CUL2FEM1B dependent ubiquitylation. Stabilization of the mitochondrial pool of FNIP1 impairs mitochondrial activity, as being read out by the oxygen consumption rate. In line with engaging endogenous FEM1B and stabilizing FNIP1, we showed that EN106 significantly reduced cellular mitochondrial oxygen consumption in HEK293T cells (Figure 2g). [1] To demonstrate that EN106 could be used as a covalent FEM1B recruiter in TPD applications, we next synthesized a series of FEM1B-based BET bromodomain degraders by linking EN106 to the BET bromodomain inhibitor JQ1 that targets BRD4 as well as other BET family proteins via 6 different linkers (Figure 3a, Figure S5). Maintaining the core benzoxazine of the alkyne probe NJH-2-030, we first attached an acetate spacer to provide methyl ester 4. The nitro group was reduced, the resulting aniline 5 mono-alkylated with acrylonitrile, and acylated to provide the chloroacetamide intermediate 6. The methyl ester was hydrolyzed under mild basic conditions and coupled to amines 7a-7f, JQ1 derivatives with different linker attachments, to provide the bifunctional degraders NJH-2-088, NJH-1-106, NJH-2-090, NJH-2-091, NJH-2-092, and NJH-2-093 (scheme for NJH-1-106 shown in Figure 3a; Figure S4). These compounds all degraded BRD4 in HEK293T cells to varying extent with NJH-1-106 showing the best degradation potency with a DC50 of 250 nM and 94 % maximal degradation of BRD4 (Figure 3b–3c, Figure S4). NJH-1-106 maintained inhibitory activity against FEM1B recognition of the FNIP1 degron with an IC50 of 1.5 μM (Figure 3d). This BRD4 degradation was time-dependent with significant degradation observed by 4 h of treatment with NJH-1-106 (Figure 3e–3f). NJH-1-106 also degraded BRD4 in cancer cell lines including the 231MFP breast cancer and HAP1 leukemia cancer cell lines (Figure S5a–S5b). [1] Consistent with the necessity of the covalent warhead, the non-reactive version of NJH-1-106, NJH-2-105, did not inhibit FEM1B interactions with the FNIP1 degron and did not degrade BRD4 (Figure S6a–S6d). Loss of BRD4 was attenuated by proteasome and NEDDylation inhibitors, consistent with a proteasome- and Cullin E3 ligase-dependent mechanism of BRD4 degradation (Figure 4a–4b). FEM1B levels remained unaltered by NJH-1-106 treatment in HEK293T cells (Figure 4a–4b). BRD4 degradation was also attenuated by pre-treatment of cells with EN106 or JQ1, showing the necessity of the ternary complex and both ends of the molecule to degrade BRD4 (Fig. 4c). In addition, BRD4 degradation was attenuated in FEM1B knockout (KO) cells compared to wild-type (WT) cells, further demonstrating FEM1B-dependent degradation of BRD4 (Fig. 4d). The incomplete rescue we observe in FEM1B KO cells could be due to residual wild-type cells in the FEM1B KO population or due to other potential off-target E3 ligases. Global proteomic profiling in HEK293T cells treated with NJH-1-106 also showed selective degradation of BRD4 amongst 4446 quantified proteins with only the largely uncharacterized PNMAL1 as an apparent off-target (Fig. 4e; Table S4). PNMAL1, however, was a likely false-positive of our proteomic analysis (Figure S7). While JQ1 is a pan BET bromodomain inhibitor, we only observed degradation of BRD4, but not BRD2 or BRD3 (Table S4). However, we cannot rule out that these other bromodomains could be degraded under longer treatments. The observed smaller fold change for BRD4 compared to the Western blot data likely reflect the well-known fold change suppression in TMT-based quantitative proteomics [1]. |
| Enzyme Assay |
Gel-Based ABPP [1]
Recombinant MBP-FEM1B1-377 (0.1μg/sample) was pre-treated with either DMSO vehicle or EN106 or at 37°C for 30 min in 25 μL of PBS, and subsequently treated with of IA-Rhodamine (concentrations designated in figure legends) at room temperature for 1 h. The reaction was stopped by addition of 4×reducing Laemmli SDS sample loading buffer. After boiling at 95°C for 5 min, the samples were separated on precast 4–20% Criterion TGX gels. Probe-labeled proteins were analyzed by in-gel fluorescence using a ChemiDoc MP. |
| Cell Assay |
Flow Cytometry Analysis of GFP-FNIP1 degron/mCherry [1]
HEK293T cells/well were seeded into 6 well plates. The next day the cells were transfected with 0.1 μg of pCS2-GFP-FNIP1562-591-IRES-mCherry or 0.1 μg of pCS2-E4F123432-GFP-IRES-mCherry, with 0.075 μg pCS2-3xFLAG-FEM1B as indicated. Empty pCS2 was added to 2 μg total DNA for each transfection in 300 μl Opti-MEM with 12 μg polyethyleneimine (PEI). Each well was transfected with 65 μl of the transfection mix. 12 hours post-transfection, indicated concentrations of EN106 or DMSO was added. After 12 hours of EN106 treatment, cells were trypsinized, spun down, resuspended in DMEM + 10% FBS and analyzed on Fortessa X20. Data was processed using FlowJo and all quantifications are the median GFP/mCherry ratios. For the pomalidomide treated cells, 10 μM pomalidomide was added for 4 hours before analyzing. |
| References | |
| Additional Infomation |
Proteolytic targeting chimeras (PROTACs) are heterobifunctional compounds composed of protein-targeting ligands linked to E3 ligase recruiters, and have become a potent therapeutic approach for targeted protein degradation (TPD). Despite the widespread application of TPD in drug development, only a handful of E3 ligase recruiters are currently available to target the more than 600 E3 ligases present in human cells. In this paper, we discovered a cysteine-reactive covalent ligand, EN106, that targets FEM1B, an E3 ligase recently discovered to be a key component in cellular responses to reductive stress. EN106 interferes with FEM1B's recognition of its key reductive stress substrate, FNIP1, by targeting the C186 site in FEM1B. We further demonstrate that EN106 can serve as a covalent recruiter for FEM1B in TPD applications. We show that PROTACs linked to EN106 with the BET bromodomain inhibitor JQ1 or the kinase inhibitor dasatinib lead to the degradation of BRD4 and BCR-ABL, respectively. Our study demonstrates a covalent ligand that targets the native E3 ligase-substrate binding site and highlights the utility of covalent ligand screening in expanding the library of E3 ligase recruiters for TPD applications. [1]
In this study, we discovered EN106, a covalent recruiter for FEM1B (an E3 ligase that controls reductive stress response), which can be used for TPD applications. While zinc has been found to be a molecular glue that links FEM1B to its endogenous substrate FNIP1, EN106 is the first synthetic small molecule ligand for FEM1B. EN106 is an early lead compound with low micromolar inhibitory potency against FEM1B and its chloroacetamide active group is metabolically unstable, requiring further medicinal chemistry studies to improve its potency, selectivity, and drug-likeness. We demonstrated that EN106 targets a cysteine residue in FEM1B that is crucial for substrate recognition. We found that EN106 can act as a FEM1B recruiter in a bifunctional degrader, recruiting potential novel substrates to the physiological target recognition sites of FEM1B, thereby enabling optimal ubiquitination via CUL2-RBX1. Surprisingly, we observed that all FEM1B-based BRD4 protacas degraded BRD4 regardless of linker length, although there was a preference for optimal Dmax and DC50 values. These data may suggest that positive cooperativity of covalent FEM1B degraders is not crucial as long as protein collisions are avoided. Future research needs to determine whether EN106 or its derivatives can act as molecular glue degraders, recruiting potential novel substrates for FEM1B-dependent ubiquitination and degradation. More broadly, future research should also focus on developing cell state-specific degraders. Furthermore, determining whether EN106 and its more effective derivatives can be used for therapy, inhibiting CUL2FEM1B and disrupting the reductive stress signaling pathway by stabilizing FNIP1 in certain cancer conditions, is also a future research direction. In summary, our study highlights the practicality of covalent ligand screening in expanding the range of E3 ligase recruiters for TPD applications. [1] |
| Molecular Formula |
C13H13CLN2O3
|
|---|---|
| Molecular Weight |
280.706922292709
|
| Exact Mass |
280.06
|
| Elemental Analysis |
C, 55.62; H, 4.67; Cl, 12.63; N, 9.98; O, 17.10
|
| CAS # |
757192-67-9
|
| PubChem CID |
5100616
|
| Appearance |
White to off-white solid powder
|
| Density |
1.351±0.06 g/cm3(Predicted)
|
| Boiling Point |
489.1±45.0 °C(Predicted)
|
| LogP |
1.3
|
| Hydrogen Bond Donor Count |
0
|
| Hydrogen Bond Acceptor Count |
4
|
| Rotatable Bond Count |
4
|
| Heavy Atom Count |
19
|
| Complexity |
368
|
| Defined Atom Stereocenter Count |
0
|
| SMILES |
ClCC(N(CCC#N)C1C=CC2=C(C=1)OCCO2)=O
|
| InChi Key |
GLDJSVHDOXCYPT-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C13H13ClN2O3/c14-9-13(17)16(5-1-4-15)10-2-3-11-12(8-10)19-7-6-18-11/h2-3,8H,1,5-7,9H2
|
| Chemical Name |
2-chloro-N-(2-cyanoethyl)-N-(2,3-dihydro-1,4-benzodioxin-6-yl)acetamide
|
| Synonyms |
EN106; EN-106; 757192-67-9; 2-chloro-N-(2-cyanoethyl)-N-(2,3-dihydro-1,4-benzodioxin-6-yl)acetamide; 2-chloro-N-(2-cyanoethyl)-N-2,3-dihydro-1,4-benzodioxin-6-ylacetamide; CHEMBL5289726; SCHEMBL23804991; DTXSID001152090; EN 106
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO : ~250 mg/mL (~890.60 mM)
|
|---|---|
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples.
Injection Formulations
Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline)(e.g. IP/IV/IM/SC) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). View More
Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] Oral Formulations
Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). View More
Oral Formulation 3: Dissolved in PEG400 (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.5624 mL | 17.8120 mL | 35.6240 mL | |
| 5 mM | 0.7125 mL | 3.5624 mL | 7.1248 mL | |
| 10 mM | 0.3562 mL | 1.7812 mL | 3.5624 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved