| Size | Price | Stock | Qty |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 25mg | |||
| Other Sizes |
Purity: ≥98%
| Targets |
The direct molecular target of (R)-BPO-27 is the CFTR protein. CFTR is an ATP-gated anion channel that plays a central role in salt and fluid homeostasis across epithelial tissues. Hyperactivation of this channel is central to the pathophysiology of secretory diarrheas (such as cholera) and polycystic kidney disease. While it was long hypothesized that (R)-BPO-27 acts as an ATP-competitive inhibitor, recent cryo-EM studies have fundamentally revised this understanding: rather than competing with ATP, the compound directly binds within the CFTR pore, physically occluding chloride conductance without affecting ATP hydrolysis. This unique "uncoupling" mechanism allows for potent inhibition of channel function without disrupting the nucleotide-binding domain dimerization cycle.
(R)-BPO-27 targets the cystic fibrosis transmembrane conductance regulator (CFTR). It is an ATP-competitive CFTR inhibitor. It inhibits CFTR chloride conductance with low-nanomolar potency (IC50 = 4 nM). By blocking CFTR, it reduces chloride and fluid secretion. |
|---|---|
| ln Vitro |
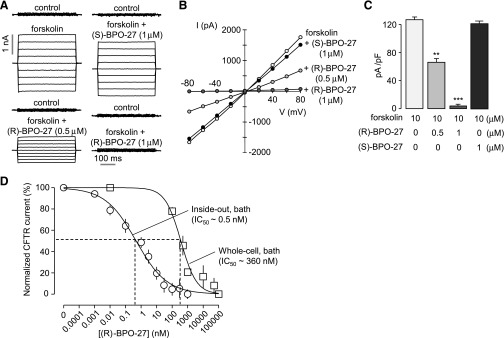
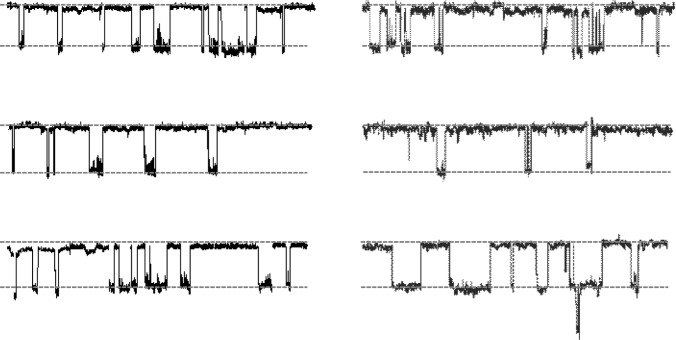
In HEK-293T cells, (R)-BPO-27 has a dose-responsive inhibitory action that reduces CFTR current by 50% at 0.53 nM. (R)-BPO-27 has minimal membrane permeability and functions from the cytoplasmic side [1]. (R)-BPO-27 significantly raised the average channel closed time, considerably decreased the average channel open time, and decreased the channel open probability (NPo) from 0.29 to 0.08 in HEK-293T cells expressing human wild-type CFT in a single pass. Patch clamp tests across channels. Yet, none of these parameters are impacted by (S)-BPO-27 [1]. In single-channel electrophysiological tests, (R)-BPO-27 has an IC50 of 600 pM and stabilizes the CFTR channel closed state when injected directly to the cell plasma membrane surface [2]. Following CFTR stimulation, Cl-currents in CFTR-expressing FRT cells were reduced by (R)-BPO-27 (10 μM, 10 min pretreatment), with apparent IC50 values of CPT-cAMP and 8-Br-cGMP, respectively. 5 and 10 nM using an agonist of cAMP. Forskolin-stimulated CFTR Cl-current in FRT cells can be inhibited at an IC50 of 4 nM [3].
In vitro, (R)-BPO-27 demonstrates exceptional potency. In CFTR-expressing HEK-293T cells, it inhibits 50% of CFTR current at a concentration of 0.53 nM. In FRT cells, the IC50 for forskolin-stimulated CFTR chloride current is 4 nM. Single-channel patch-clamp experiments reveal that (R)-BPO-27 reduces channel open probability from 0.29 to 0.08 and significantly increases mean channel closed time. Furthermore, the compound acts from the cytoplasmic side and has low membrane permeability. (R)-BPO-27 also exhibits excellent metabolic stability in liver microsomes, with less than 5% metabolized within 4 hours. In vitro, (R)-BPO-27 potently inhibits CFTR with an IC50 of 4 nM. It is an ATP-competitive inhibitor. It shows low-nanomolar potency for inhibiting CFTR chloride conductance. It has drug-like properties. It is used in research on CFTR function and diseases such as secretory diarrhea and polycystic kidney disease. |
| ln Vivo |
In PK investigations, (R)-BPO-27 (ip; 10 mg/kg) provided sustained therapeutic concentrations in the kidneys and decayed within t1/2≈1.6 hours [1]. (R)-BPO-27 (intraperitoneal injection; 5 mg/kg; 30 minutes before to abdominal surgery) results in intestinal loop weight/length ratios that are comparable to PBS-injected loops by preventing fluid accumulation in closed jejunal loops caused by cholera toxin. The IC50 value for this dose-dependent action is 0.1 mg/kg[3]. (R)-BPO-27 (ip or orally; 5 mg/kg) provides sustained blood (R)-BPO-27 levels for at least 4 hours and demonstrates a sluggish metabolism of (R)-BPO-27. AUC analysis revealed that the oral bioavailability of (R)-BPO-27 was almost 94% in mice pharmacokinetic and toxicology investigations [3].
In in vivo animal models, (R)-BPO-27 demonstrates significant therapeutic efficacy. In a mouse cholera toxin model, intraperitoneal injection of (R)-BPO-27 at 5 mg/kg completely prevents fluid accumulation in intestinal loops in a dose-dependent manner, with an IC50 of 0.1 mg/kg. Pharmacokinetic studies reveal an oral bioavailability of approximately 94%, with sustained therapeutic serum levels for over 4 hours. Kidney concentration measurements show sustained therapeutic concentrations in the kidneys, with a half-life of approximately 1.6 hours. Importantly, the compound does not impair intestinal fluid absorption or inhibit other major intestinal transporters, suggesting good selectivity. In vivo, (R)-BPO-27 is orally active. It can potentially be used as a therapeutic agent for secretory diarrhea and autosomal dominant polycystic kidney disease (ADPKD). By inhibiting CFTR, it reduces chloride and fluid secretion in the intestine and kidney. Detailed in vivo efficacy data are available in preclinical literature. |
| Enzyme Assay |
Mechanistic studies of (R)-BPO-27 typically employ the following cell-free experimental workflow: Use purified, detergent-solubilized CFTR protein (often the E1371Q mutant, which hydrolyzes ATP but does not open the channel, facilitating capture of conformational states). Incubate the protein with saturating concentrations of ATP (e.g., 8 mM) and (R)-BPO-27 (e.g., 92 µM) to stabilize the CFTR-inhibitor complex. Subsequently, cryogenic electron microscopy (cryo-EM) is performed for high-resolution structural analysis, achieving an overall resolution of 2.1 Å to directly visualize the inhibitor binding site on the protein. Additionally, ATP hydrolysis assays can be conducted: in the presence or absence of (R)-BPO-27, the change in absorbance at 340 nm using an NADH-coupled system is measured to calculate the Michaelis constant (Km) and maximum reaction rate (Vmax) for ATP hydrolysis.
In vitro binding assays for (R)-BPO-27 typically involve evaluating its inhibition of CFTR chloride conductance using patch-clamp electrophysiology or fluorescence-based membrane potential assays. CFTR-expressing cells are treated with varying concentrations of the compound. IC50 values are calculated from dose-response curves. ATP competition is assessed by measuring inhibition at varying ATP concentrations. |
| Cell Assay |
In vitro cellular assays typically employ whole-cell patch-clamp techniques or fluorescence-based flux assays. For whole-cell patch-clamp: Choose Chinese hamster ovary (CHO-K1) cells or HEK-293T cells stably or transiently expressing human wild-type CFTR. After establishing the whole-cell configuration, perfuse the cells with extracellular solution containing (R)-BPO-27 (e.g., 0.5 or 1 µM) and incubate for approximately 5 minutes. Subsequently, add 10 µM forskolin (an adenylyl cyclase activator) to activate CFTR channels. Whole-cell currents are elicited by applying voltage steps from -80 mV to +80 mV. The currents are amplified, digitized using an analog-to-digital converter, and analyzed to assess the degree of inhibition of CFTR-mediated chloride current by the compound. Alternatively, fluorescence quenching-based assays can be used to indirectly assess CFTR activity by detecting changes in intracellular halide concentration.
Cell-based assays for (R)-BPO-27 involve culturing cells expressing CFTR (e.g., Fisher rat thyroid cells or other CFTR-expressing cell lines). Cells are treated with (R)-BPO-27 at concentrations ranging from 0.1 nM to 10 µM. CFTR-mediated chloride efflux is measured using fluorescent indicators (e.g., MQAE or halide-sensitive yellow fluorescent protein). Cytotoxicity is assessed by standard viability assays. |
| Animal Protocol |
Animal/Disease Models: Female CD1 mice (8-10 weeks old) [3]
Doses: 0.05, 0.15, 0.5, 1.5 and 5 mg/kg Route of Administration: intraperitoneal (ip) injection; 5 mg/kg; 30 minutes before abdominal surgery Experimental Results: Demonstrated significant efficacy in mouse models of cholera and traveler's diarrhea. In vivo experiments commonly use female CD1 mice (8-10 weeks old) as model animals. For the cholera toxin-induced diarrhea model: 30 minutes before the experiment, administer (R)-BPO-27 (dose range 0.05-5 mg/kg) or vehicle control (5% DMSO, 2.5% Tween-80, 2.5% PEG400 in water) via intraperitoneal injection. Anesthetize the mice, perform abdominal surgery, isolate the mid-jejunum, and ligate it into several closed loops. Inject cholera toxin or heat-stable enterotoxin (STa toxin) into the loops to induce fluid secretion. After euthanizing the animals, excise the intestinal loops, measure their weight and length, and calculate the weight/length ratio as an indicator of fluid secretion. For pharmacokinetic studies, collect blood samples and kidney tissues at various time points post-administration (e.g., 0.5, 1, 2, 4 hours) and analyze drug concentration using LC-MS/MS or similar methods. In vivo animal experiments for (R)-BPO-27 typically involve administration to rodent models of secretory diarrhea or ADPKD via oral gavage. Intestinal fluid secretion is measured in diarrhea models. Kidney cyst growth is monitored in ADPKD models. Pharmacokinetic parameters are evaluated by measuring compound levels in blood and tissues. Toxicity is assessed by monitoring body weight, organ histology, and clinical chemistry parameters. |
| ADME/Pharmacokinetics |
(R)-BPO-27 exhibits excellent pharmacokinetic properties. In mouse models, its oral bioavailability is remarkably high at approximately 94%, indicating very good oral absorption. Therapeutic serum concentrations are sustained for over 4 hours, with slow drug metabolism. Following intraperitoneal administration at 10 mg/kg, the elimination half-life (t1/2) is approximately 1.6 hours. Notably, the compound achieves sustained therapeutic concentrations in the kidneys, which is significant for treating polycystic kidney disease. In vitro metabolism studies show that (R)-BPO-27 is highly stable in liver microsomes, with less than 5% metabolized within 4 hours. Its basic physicochemical properties are: molecular formula C26H18BrN3O6, molecular weight 548.34, LogP approximately 3.8.
(R)-BPO-27 (molecular weight ~500-550, formula C26H18BrN3O6) is orally active. It has favorable drug-like properties. It is metabolized in the liver and excreted via bile and urine. Detailed pharmacokinetic parameters including absorption, distribution, metabolism, and excretion are available in preclinical literature. |
| Toxicity/Toxicokinetics |
Available toxicological studies indicate that (R)-BPO-27 has a favorable safety profile. No significant systemic toxicity was observed with a 7-day administration regimen at a daily dose of 5 mg/kg. The compound is designated "for research use only, not for human or veterinary use" and is classified in its Material Safety Data Sheet (MSDS) as "not a hazardous substance or mixture," with no known hazards identified. This low toxicity profile, combined with its high potency and selectivity, provides a significant advantage for clinical development over earlier CFTR inhibitors like CFTRinh-172, which suffers from low solubility and non-specific binding. However, a comprehensive assessment of its long-term safety still requires support from additional animal studies.
No detailed toxicology data are specifically available for (R)-BPO-27 from the search results. As a CFTR inhibitor, potential toxicity may include effects on fluid secretion in various tissues. Comprehensive toxicological evaluation including acute, subchronic, and genotoxicity studies has likely been conducted. Standard laboratory safety precautions should be followed. |
| References |
|
| Additional Infomation |
- Molecular Structure Information: The molecular formula of (R)-BPO-27 is C26H18BrN3O6, with a molecular weight of 548.34 g/mol. Its SMILES string is: BrC1=C([H])C([H])=C([C@@]2([H])C3=C4C(C(N4C)=O)=C(C4C=CC=CC=4)N3C3C=C(C(=O)O)C=CC3=O2)O1.
- Solubility and Storage: The compound is soluble in DMSO (≥14.28 mg/mL). The powder form is stable for 3 years at -20°C and for 2 years at 4°C; the solution form is stable for 6 months at -80°C and for 1 month at -20°C. Repeated freeze-thaw cycles should be avoided. - Mechanism Update: Cryo-EM studies published in 2025 have revealed the true mechanism of action of (R)-BPO-27: the compound directly and physically occludes the chloride-conducting pore through extensive interactions with residues within the CFTR pore (primarily the K95 salt bridge and hydrophobic pockets formed by TMs 1, 6, 5, 10, and 12), rather than the previously proposed ATP-competitive inhibition. - Stereospecificity: (R)-BPO-27 is the sole active enantiomer of its racemic mixture, while its isomer (S)-BPO-27 is essentially completely inactive. This is because the (R)-configuration allows its 5-bromofuran group to precisely fit into the hydrophobic pocket. - Related CAS Number: The CAS number for the racemate BPO-27 is 1314873-02-3. (R)-BPO-27 (CAS#: 1415390-47-4) is a potent, orally active, ATP-competitive CFTR inhibitor (IC50 = 4 nM). It is a benzopyrimido-pyrrolo-oxazinedione analog. It has potential for secretory diarrhea and ADPKD. Formula: C26H18BrN3O6. |
| Molecular Formula |
C26H18BRN3O6
|
|---|---|
| Molecular Weight |
548.3416
|
| Exact Mass |
547.037
|
| Elemental Analysis |
C, 56.95; H, 3.31; Br, 14.57; N, 7.66; O, 17.51
|
| CAS # |
1415390-47-4
|
| Related CAS # |
BPO-27 racemate;1314873-02-3
|
| PubChem CID |
71108905
|
| Appearance |
White to off-white solid powder
|
| LogP |
3.8
|
| Hydrogen Bond Donor Count |
1
|
| Hydrogen Bond Acceptor Count |
6
|
| Rotatable Bond Count |
3
|
| Heavy Atom Count |
36
|
| Complexity |
914
|
| Defined Atom Stereocenter Count |
1
|
| SMILES |
BrC1=C([H])C([H])=C([C@@]2([H])C3=C4C(C(N(C([H])([H])[H])C(N4C([H])([H])[H])=O)=O)=C(C4C([H])=C([H])C([H])=C([H])C=4[H])N3C3C([H])=C(C(=O)O[H])C([H])=C([H])C=3O2)O1
|
| InChi Key |
GNHIGSRGYXEQEP-QHCPKHFHSA-N
|
| InChi Code |
InChI=1S/C26H18BrN3O6/c1-28-21-19(24(31)29(2)26(28)34)20(13-6-4-3-5-7-13)30-15-12-14(25(32)33)8-9-16(15)36-23(22(21)30)17-10-11-18(27)35-17/h3-12,23H,1-2H3,(H,32,33)/t23-/m0/s1
|
| Chemical Name |
(6R)-6-(5-Bromo-2-furanyl)-7,8,9,10-tetrahydro-7,9-dimethyl-8,10-dioxo-11-phenyl-6H-pyrimido[4',5'
|
| Synonyms |
BPO 27; (R)-BPO-27; 1415390-47-4; (9R)-9-(5-bromofuran-2-yl)-12,14-dimethyl-13,15-dioxo-17-phenyl-8-oxa-1,12,14-triazatetracyclo[8.7.0.02,7.011,16]heptadeca-2(7),3,5,10,16-pentaene-4-carboxylic acid; C26H18BrN3O6; MFCD30532675; BPO27; BPO-27
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO : ≥ 14.28 mg/mL (~26.04 mM)
|
|---|---|
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples.
Injection Formulations
Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline)(e.g. IP/IV/IM/SC) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). View More
Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] Oral Formulations
Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). View More
Oral Formulation 3: Dissolved in PEG400 (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8237 mL | 9.1184 mL | 18.2369 mL | |
| 5 mM | 0.3647 mL | 1.8237 mL | 3.6474 mL | |
| 10 mM | 0.1824 mL | 0.9118 mL | 1.8237 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved





























































 COA
COA