| Size | Price | Stock | Qty |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg | |||
| Other Sizes |
| Targets |
mGluR7
|
|---|---|
| ln Vitro |
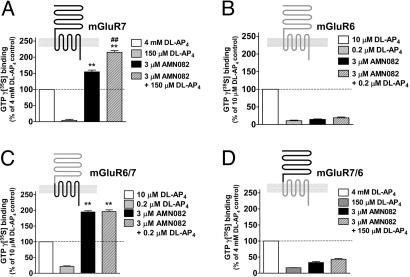
Without altering the baseline glutamate release, preincubating synaptosomes with AMN082 (1 μM) for 10 min prior to 4-aminopyridine treatment efficiently suppresses 4-aminopyridine-evoked glutamate release [2].
\n\nAMN082 is a selective metabotropic glutamate mGlu7 receptor agonist reported to exhibit antidepressant activity. Considering that excessive glutamate release is involved in the pathogenesis of depression, the effect of N,N'-dibenzyhydryl-ethane-1,2-diamine dihydrochloride (AMN082) on glutamate release in rat cerebrocortical nerve terminals and the possible underlying mechanism were investigated. In this study, we observed here that AMN082 inhibited 4-aminopyridine-evoked glutamate release and this phenomenon was blocked by the metabotropic glutamate mGlu7 receptor antagonist MMPIP. Moreover, western blot analysis and immunocytochemistry confirmed the presence of presynaptic metabotropic glutamate mGlu7 receptor proteins. The effect of AMN082 on the 4-aminopyridine-evoked release of glutamate was prevented by chelating the extracellular Ca2+ ions and the vesicular transporter inhibitor; however, the effect of AMN082 was unaffected by the glutamate transporter inhibitor. AMN082 reduced the elevation of 4-aminopyridine-evoked intrasynaptosomal Ca2+ concentration, but did not alter the synaptosomal membrane potential. In the presence of the Cav2.2 (N-type) and Cav2.1 (P/Q-type) channel blocker, the adenylate cyclase inhibitor, and the protein kinase A inhibitor, the action of AMN082 on the 4-aminopyridine-evoked glutamate release was markedly reduced. These results suggest that the activation of the metabotropic glutamate mGlu7 receptors by AMN-082 reduces adenylate cyclase/protein kinase A activation, which subsequently reduces the entry of Ca2+ through voltage-dependent Ca2+ channels and decreases evoked glutamate release. Additionally, fluoxetine, a clinically effective antidepressant, completely occluded the inhibitory effect of AMN082 on glutamate release, thus indicating the existence of a common intracellular mechanism for these two compounds to inhibit glutamate release from the cerebrocortical nerve terminals. [2]\n \nMetabotropic glutamate receptor (mGluR) subtypes (mGluR1 to mGluR8) act as important pre- and postsynaptic regulators of neurotransmission in the CNS. These receptors consist of two domains, an extracellular region containing the orthosteric agonist site and a transmembrane heptahelical domain involved in G protein activation and recognition of several recently synthesized pharmacological modulators. The presynaptic receptor mGluR7 shows the highest evolutionary conservation within the family, but no selective pharmacological tool was known. Here we characterize an mGluR7-selective agonist, N,N'-dibenzhydrylethane-1,2-diamine dihydrochloride (AMN082), which directly activates receptor signaling via an allosteric site in the transmembrane domain. At transfected mammalian cells expressing mGluR7, AMN082 potently inhibits cAMP accumulation and stimulates GTPgammaS binding (EC50-values, 64-290 nM) with agonist efficacies comparable with those of L-2-amino-4-phosphonobutyrate (L-AP4) and superior to those of L-glutamate. AMN082 (< or = 10 microM) failed to show appreciable activating or inhibitory effects at other mGluR subtypes and selected ionotropic GluRs. Chimeric receptor studies position the binding site of AMN082 in the transmembrane region of mGluR7, and we demonstrate that this allosteric agonist has little, if any, effect on the potency of orthosteric ligands. Here we provide evidence for full agonist activity mediated by the heptahelical domain of family 3 G protein-coupled receptors (which have mGluR-like structure) that may lead to drug development opportunities. [1] \n\nEffect of AMN082 on Cloned mGluR7. AMN082 was identified by using high-throughput random screening of chemical libraries. The chemical structure of AMN082 (Fig. 1) is completely unrelated to the known mGluR7 ligands, which are all derived from the l-glutamate backbone. This compound elicited a concentration-dependent inhibition of forskolin-stimulated cAMP accumulation in CHO cells stably expressing human mGluR7b (Fig. 1 A; EC50, 64 ± 32 nM), comparable in efficacy with a saturating concentration of the orthosteric group III mGluR agonist DL-AP4 (Fig. 1A, dotted line). Up to 3 μM, there was no effect of AMN082 in the same assay conducted with mGluR2-expressing CHO cells (Fig. 1B). Stimulation of GTPγ35S binding was performed to evaluate further AMN082's pharmacological properties and mechanism of action. AMN082 (3 μM) produced 167 ± 8% stimulation relative to the maximal agonist activity of l-glutamate (set to 100%; Fig. 1C). The stimulating effects of AMN082 were almost additive with those of l-glutamate and DL-AP4 (238 ± 11% and 336 ± 19%, respectively), whereas DL-AP4 plus l-glutamate (both at maximally active concentrations) produced a smaller stimulation than DL-AP4 alone (133 ± 4% vs. 216 ± 12%, Fig. 1C). Thus, l-glutamate and DL-AP4 are likely to interact at the same receptor site, and the activity of the full agonist DL-AP4 seems to be inhibited by the partial agonist l-glutamate. Next, the group III mGluR-selective antagonists MSOP and CPPG were tested against concentration-response curves of AMN082 conducted in the presence of submaximal DL-AP4 (Fig. 1D): the DL-AP4 component was completely abolished by the antagonists, whereas there was no inhibition of the AMN082-stimulated GTPγ35S binding. Together, the data of Fig. 1 C and D suggest that AMN082 activates mGluR7 signaling most likely by binding to a different site than the orthosteric ligands l-glutamate, DL-AP4, MSOP, and CPPG. [1] \n\nConcentration-response curves for AMN082, DL-AP4, and l-glutamate are compared in Fig. 2A. AMN082 is far more potent than the orthosteric ligands, DL-AP4 and l-glutamate; the EC50 values (95% confidence intervals) are 260 nM (200; 360), 540 μM (440; 670), and 700 μM (580; 850), respectively. To analyze a potential cooperativity between l-glutamate site ligands and AMN082, concentration-response curves for AMN082 at different fixed concentrations of l-glutamate and, inversely, curves for l-glutamate vs. fixed concentrations of AMN082 were conducted (Fig. 2 B-D). The EC50 of AMN082 varied between 140 and 290 nM with largely overlapping 95% confidence intervals (Fig. 2 B and D). Similarly, the EC50 of l-glutamate was consistently between 640 and 830 μM, irrespective of the added concentration of AMN082 (Fig. 2 C and D). To address whether binding of AMN082 to mGluR7 affects the binding affinity of ligands for the l-glutamate site, we analyzed displacement of 10 nM [3H]LY341495 (a competitive mGluR antagonist) binding from membranes prepared from CHO cells stably expressing mGluR7a; up to 30 μM AMN082 showed no displacement of this radioligand. In contrast, 10 mM L-AP4, l-glutamate, or L-SOP abolished 100% of specific binding (data not shown). Moreover, we conducted [3H]LY341495 displacement curves with L-SOP, L-AP4, and l-glutamate, each in the absence and presence of 3 μM AMN082. The addition of AMN082 induced only a minor left-shift of each of the three curves; the calculated Ki values with 95% confidence intervals (in parentheses in μM) were as follows: L-SOP, 45 μM (39; 50); L-SOP plus 3 μM AMN082, 29 μM (24; 35); L-AP4, 193 μM (165; 224); L-AP4 plus 3 μM AMN082, 176 μM (144; 216); l-glutamate, 624 μM (446; 870); and l-glutamate plus 3 μM AMN082, 524 μM (436; 631) (data not shown). [1] \n\nAMN082 Directly Interacts with the Heptahelical Region of mGluR7. Next, we intended to localize the binding site of AMN082 to one discrete region of the mGluR7 protein and decided to use constructs of wild-type mGluR7b and mGluR6 as well as two chimeras: the mGluR6/7b construct contains the N-terminal extracellular region of mGluR6 and the C-terminal portion of mGluR7b comprising the entire transmembrane region; mGluR7/6 is the reverse chimera (see Materials and Methods and Fig. 3). When using the stimulation of GTPγ35S binding, the activity of AMN082 on mGluR6/7b and mGluR7/6 chimeras was very similar to wild-type mGluR7b and mGluR6, respectively: AMN082 stimulated mGluR7b and mGluR6/7b by 150-200% relative to the maximal effect of DL-AP4, but AMN082 produced only minor effects on mGluR6- and mGluR7/6-expressing membranes (10-25% relative to maximal DL-AP4 stimulation) (Fig. 3). It is interesting to note that the stimulating effects of AMN082 in combination with DL-AP4 were more than just additive on those mGluR7-expressing, but not on mGluR6/7b-expressing, membranes (Fig. 3 A and C). In addition, initial attempts using truncated mutants with deleted extracellular domains were made, but no activation with DL-AP4 or AMN082 was observed (data not shown); it cannot be ruled out that their translated proteins were misfolded or incorrectly inserted into membranes. [1] \n\nSelectivity Profiling of AMN082. Before we addressed the activity of AMN082 at all eight known mGluRs and at three selected ionotropic receptors, we confirmed that there was no significant binding interaction of 1 μM AMN082 with 30 different nervous system targets using a radioligand displacement assay; this list included a selection of receptors for adrenaline, dopamine, GABA, histamine, acetylcholine, opiates, serotonin, and substance P plus selected neurotransmitter reuptake sites (n = 2-4 determinations per target; data not shown). Fig. 4 shows the effects of AMN082 on all eight mGluRs and on three ionotropic GluRs. The activating effect of 3 μM and 10 μM AMN082 was selectively seen at mGluR7a and mGluR7b with large efficacies of 70-140% (relative to maximal DL-AP4 effects) when using the stimulation of GTPγ35S binding (Fig. 4 A and D). Under an identical assay design, AMN082 (up to 10 μM) elicited little or no stimulating effects on membranes from mGluR2-, mGluR3-, mGluR4-, mGluR6-, or mGluR8a-expressing cells, and there was also no activation of GTPγ35S binding in untransfected CHO cells (Fig. 4 B-D). Measurements of phosphoinositol hydrolysis were done to address whether AMN082 activates group I mGluR subtypes. AMN082 displays neither agonist-like nor positive modulatory activity at mGluR1b- or mGluR5a-expressing cells (Fig. 4D). Functional agonist and modulatory activities of AMN082 were also excluded for two NMDAR subtypes plus one α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subtype (NMDAR1a/2A, NMDAR1a/2B, and GluR3; Fig. 4D); this test was done via cytoplasmic calcium determinations by using stably transfected cell lines. Furthermore, up to 10 μM, AMN082 showed no antagonist-like effects at any of the tested mGluR or ionotropic GluR subtypes when using the functional receptor assay formats described above (Fig. 4). [1] \n\n\n\n |
| ln Vivo |
AMN082 (6 mg/kg; oral) generates a rise in stress hormones in a mGluR7+/+ mouse (C57BL/6 genetic background) in a mGluR7-dependent way [1]. AMN082 (1.25-5.0 mg/kg, i.p.; 30 minutes before each injection of cocaine or morphine during repeated dosing or before cocaine or morphine challenge) dose-dependently attenuates the development and expression of cocaine or morphine locomotor sensitization [3] .\n
\n\nIn Vivo Activity of AMN082: Modulation of Stress Hormones in an mGluR7-Dependent Fashion. Unlike all known l-glutamate site ligands for group III mGluRs, AMN082 readily passes the blood-brain barrier upon oral administration: 10 mg/kg oral AMN082 results in 0.29 μmol/kg in total brain tissue, and 14 mg/kg leads to 0.62 μmol/kg in rats and mice, respectively, 1 hour after oral administration (data not shown).\n\nThe role of mGluR7 in stress-related behavior is well documented. Therefore, we analyzed the effect of oral administration of AMN082 on serum levels of the stress hormones corticosterone and ACTH. Fig. 5A shows that AMN082 increases plasma corticosterone in a dose-dependent manner in a wild-type mouse strain (C57BL/6). Next, we used mGluR7-deficient mice (mGluR7-/-) and their wild-type littermates (mGluR7+/+). Again, oral administration of 6 mg/kg AMN082 elicited an increase of ≈200% of plasma corticosterone in mGluR7+/+ animals, but no such rise was observed in mGluR7-/- mice (Fig. 5B). Similarly, blood levels of ACTH were also increased to 200% 1 hour after oral AMN082 administration in wild-type animals, but not in mGluR7-deficient mice (Fig. 5C).[1] \n\nPrevious studies have indicated that metabotropic glutamate receptors 7 (mGluR7s) are involved in drug addiction. However, the role of these receptors in drug-induced behavioral sensitization is unknown. The aim of the present study was to determine whether systemic injection of AMN082, a selective mGluR7 allosteric agonist, reduces the cocaine- and morphine-induced hyperactivity and the development and expression of locomotor sensitization, and also affects the reciprocal cross-sensitization to the stimulant effect of cocaine and morphine in mice. AMN082 (1.25-10.0 mg/kg, i.p.) did not have an impact on locomotion of naive mice and did not affect the acute cocaine- or morphine-induced hyperactivity, except the dose of 10 mg/kg that suppressed the locomotor effect of both drugs. Repeated exposure to cocaine or morphine (10 mg/kg, 5× every 3 days) gradually increased locomotion during induction of sensitization and after 4 (cocaine) or 7 day (morphine) withdrawal phase when challenged with cocaine (10 mg/kg, i.p.) or morphine (10 mg/kg, i.p.) on day 17 or 20, respectively. Pretreatment of animals with the lower doses of AMN082 (1.25-5.0 mg/kg, i.p.), 30 min before every cocaine or morphine injection during repeated drug administration or before cocaine or morphine challenge, dose-dependently attenuated the development, as well as the expression of cocaine or morphine locomotor sensitization. AMN082 also inhibited the reciprocal cross-sensitization between these drugs. Prior to administration of MMPIP (10 mg/kg, i.p.), a selective mGluR7 antagonist reversed the inhibitory effect of AMN082 on the development or expression of cocaine or morphine sensitization. These data indicate that AMN082 attenuated the development and expression of cocaine and morphine sensitization, and the reciprocal cross-sensitization via a mechanism that involves mGluR7s. Thus, AMN082 might have therapeutic implications not only in the treatment of cocaine or opioid addiction but also in the treatment of cocaine/opioid polydrug-abusers. [3]\n \nEffect of AMN082 treatment on the acute cocaine- or morphine-induced hyperlocomotion and basal locomotor activity.\nEffect of co-administration of AMN082 on the development of sensitization to the locomotor stimulant effect of cocaine and morphine. Influence of MMPIP on the AMN082 effect.\nEffect of AMN082 treatment on the expression of sensitization to the locomotor stimulant effect of cocaine and morphine. Influence of MMPIP on the AMN082 effect.\nEffect of AMN082 treatment on the expression of reciprocal locomotor cross-sensitization between cocaine and morphine. [3] |
| Enzyme Assay |
GTPγ35S Binding Assays. [1]
Membranes were prepared from transfected mGluR2-, mGluR3-, mGluR4-, mGluR6-, mGluR7a-, mGluR7b-, mGluR8a-, mGluR6/7b-, and mGluR7/6-expressing cells, and GTPγ35S binding assays were conducted by using the protocol described by Maj et al. Second-Messenger Assays. [1] Measurements of cAMP accumulation were performed as previously described by using CHO cell lines stably expressing individual mGluR subtypes. Measurement of [3H]inositol phosphate formation was done according to Gasparini et al., and calcium measurements were done as described by Maj et al. [3H]LY341495 Binding Assay. [1] Membrane fractions of CHO cells stably expressing mGluR7a (see above) were diluted in assay buffer (10 mM KH2PO4/100 mM KBr, pH 7.6), homogenized briefly by using a Polytron homogenizer, and incubated for 10 min at 30°C. Assay mixtures were prepared in 96-well microtiter plates. The composition of the assay mixtures in a final volume of 200 μl per well was as follows: 10 mM KH2PO4/100 mM KBr (pH 7.6), 50 μg of pretreated membrane protein, 1.5 mg of wheat germ agglutinin scintillation proximity assay (WGA SPA) beads, 10 nM [3H]LY341495, and the test compounds at the appropriate concentrations. Nonspecific binding was measured in the presence of 1 mM l-serine-O-phosphate (L-SOP). The samples were incubated for 60 min at room temperature (with shaking), before being counted in a TopCount. Data were analyzed by using nonlinear regression in the prism program. IC50 values were converted into Ki values by using the Cheng and Prusoff equation. |
| Cell Assay |
Glutamate release [2]
Glutamate release was assayed by on-line fluorometry (Nicholls and Sihra, 1986, Lin et al., 2015). Pelleted synaptosomes were resuspended in HEPES buffer medium containing 16 μM bovine serum albumin and incubated in a stirred and thermostated cuvette at 370C in a Perkin-Elmer LS-55 spectrofluorimeter. NADP+ (2 mM), glutamate dehydrogenase (50 units/ml) and CaCl2 (1.2 mM) were added after 3 min 4-aminopyridine (1 mM) was applied to evoked glutamate release. The oxidative deamination of released glutamate, leading to the reduction of NADP+, was monitored by measuring NADPH fluorescence at excitation and emission wavelengths of 340 and 460 nm, respectively. A standard of exogenous glutamate (5 nmol) was added at the end of each experiment and the fluorescence change produced by the standard addition was used to calculate the released glutamate as nanomoles glutamate per milligram synaptosomal protein (nmol/mg). Release values quoted in the text and depicted in bar graphs represent the levels of glutamate cumulatively released after 5 min of depolarization, and are expressed as nmol/mg/5 min. Data were accumulated at 2-s intervals and cumulative data were analyzed using Lotus 1–2-3. The cytosolic free Ca2+ concentration ([Ca2+]C) in the synaptosomal population [2] [Ca2+]C was measured with fura-2. Synaptosomes were resuspended (2 mg/ml) in HEPES buffer medium containing 16 μM bovine serum albumin in the presence of 5 μM fura-2 and 0.1 mM CaCl2 and incubated at 37 °C for 30 min in a stirred test tube. After fura-2 loading, synaptosomes were pelleted and resuspended in HEPES buffer medium containing bovine serum albumin. The synaptosomal suspension was stirred in a thermostatted cuvette containing 1.2 mM CaCl2 in a Perkin-Elmer LS-55 spectrofluorimeter, and the fluorescence was monitored at excitation wavelengths of 340 and 380 nm (emission wavelength 505 nm). Data was collected at 2-s intervals and the [Ca2+]C (nM) was calculated using the equations described previously (Grynkiewicz et al., 1985). Western blotting [2] Synaptosomes were lysed in ice-cold Tris-buffered solution (50 mM Tris/HCl, 150 mM NaCl, 1% Triton X-100, protease inhibitor cocktail, pH 7.4) and quantified for protein content with protein assay kit. Then, proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes and incubated overnight at 40C with one of the following rabbit monoclonal antibodies: metabotropic glutamate mGlu7 (1:500) and β-actin (1:2000). After washing, the membranes were incubated for 1 h with peroxidase-conjugated goat anti-rabbit secondary antibodies (1:50,00). Immunoreactivity was detected using an enhanced chemiluminescence and quantitative densitometry using the Syngene software Values in the figure are the mean of at least three independent experiments. Immunocytochemistry [2] The synaptosomes were placed onto the polylysine-coated coverslips (diameter 20 mm) for 1 h, fixed with 4% paraformaldehyde in 0.1 M phosphate-buffered saline for 15 min, permeabilized with 0.2% Triton X-100 phosphate-buffered saline for 5 min and incubated for 24 h with the appropriate primary antibody against vesicular transporter of glutamate type 1 (1:200) or mGlu7 receptor (1:200), diluted in 50 mM Tris buffer containing 0.9% NaCl with 0.2% Triton X-100. After washing with Tris buffer containing 0.9% NaCl, the synaptosomes were incubated with secondary antibodies diluted in Tris buffer containing 0.9% NaCl for 1 h: goat anti-mouse DyLight 549 (red; 1:200) or goat anti-rabbit FITG (green; 1:200). After several washes in Tris buffer containing 0.9% NaCl, the coverslips were mounted with fluoromount and the synaptosomes were viewed with a magnification of 400×, using upright fluorescence microscopy, and images were captured using a CCD camera. Each coverslip was analyzed by counting at least three different fields. |
| Animal Protocol |
Animal/Disease Models: Male Swiss mice (20-25g) [3]
Doses: 1.25, 2.5, 5.0 mg/kg Route of Administration: intraperitoneal (ip) injection; on day 17 or day 20, cocaine (10 mg/kg) or Results of morphine (10 mg/kg) given 30 minutes before challenge: Dramatically attenuated the expression of cocaine-induced locomotor sensitization; attenuated morphine-induced sensitization. Animal Procedures, in Vivo AMN082 Administration. mGluR7-/- mice were generated as described from E14 (129/Ola) embryonic stem cells. All of the mice in the studies reported here carried wild-type or mutant mGluR7 alleles on a 14th-generation (F14) C57BL/6 genetic background. Age-matched groups of mGluR7-/- and mGluR7+/+ mice were generated as described. Male animals were used in all experiments. Food pellets and tap water were available ad libitum. Male mGluR7+/+ and littermate mGluR7-/- mice were injected orally (p.o.) with vehicle or 1-6 mg/kg AMN082. One hour later, mice (mGluR7-/- and mGluR7+/+ mice in a randomized order) were decapitated rapidly (within 30 sec after first touching the cage), and trunk blood was collected (n ≥ 9 per genotype). All animal experiments were subject to institutional review and conducted in accordance with the Veterinary Authority of Basel-Stadt. For i.p. injection, AMN082 and MMPIP were suspended in 0.5% methylcellulose, which was used as a vehicle. Both compounds were administered in a volume of 10 ml/kg (0.01 ml/g body weight). Fresh drug solutions were prepared on each day of the experiments. Drug injection times were determined based upon pilot studies and literature reports. [3] Effect of AMN082 treatment on the acute cocaine- or morphine-induced hyperlocomotion and basal locomotor activity [3] Mice (n = 6–10) were pretreated with a single injection of AMN082 (1.25, 2.5, 5.0 or 10 mg/kg, i.p.) or vehicle 30 min before injection of cocaine (10 mg/kg, i.p.) or morphine (10 mg/kg, i.p.). Locomotor activity was recorded for 30 or 60 min, respectively, immediately after cocaine/morphine administration. Furthermore, an influence of AMN082 (1.25, 2.5, 5.0 and 10 mg/kg, i.p.) alone on the locomotor activity of naive mice was examined. Effect of co-administration of AMN082 on the development of sensitization to the locomotor stimulant effect of cocaine and morphine. Influence of MMPIP on the AMN082 effect [3] To investigate the influence of AMN082 on the development of sensitization to the locomotor stimulant effect of cocaine, the mice (n = 7–9) were pretreated with AMN082 (1.2, 2.5 or 5.0 mg/kg, i.p.) or vehicle, 30 min before cocaine (10 mg/kg, i.p.) or saline (control group) injection on the first day (day 1) of the experiment. The mice were directly placed in the test apparatus and locomotor activity was measured for the following 30 min. The administration procedure was repeated on days 4, 7, 10, and 13, and locomotor activity measurements were performed as described above. To determine whether mGluR7s are involved in the AMN082 effect on the development of cocaine sensitization, the cocaine-treated group was pretreated with MMPIP (10 mg/kg, i.p.), an allosteric mGluR7-selective antagonist, 30 min prior to AMN082 (5.0 mg/kg, i.p.) injection. Control mice were injected with saline and vehicle. Following a withdrawal period of 4 days (day 17), all groups of mice were challenged with cocaine (10 mg/kg, i.p.) without AMN082 and/or MMPIP administration and the locomotor activity was measured for 30 min. To investigate the influence of AMN082 on the development of sensitization to the locomotor stimulant effect of morphine, the mice (n = 6–10) were pretreated with AMN082 (1.25, 2.5 or 5.0 mg/kg, i.p.) or vehicle, 30 min before each morphine (10 mg/kg, i.p.) or saline (control group) injection every 3 days, for five times (on the 1st, 4th, 7th, 10th and 13th days). Immediately after morphine administration, locomotor activity was measured for 60 min. To determine whether the mGluR7s are involved in the AMN082 effect on the induction of morphine locomotor sensitization, the morphine-treated group was pretreated with MMPIP (10 mg/kg, i.p.), 30 min prior to AMN082 (5.0 mg/kg, i.p.) injection. Control mice were injected with saline and vehicle. Following a withdrawal period of 7 days (day 20), all groups of mice received a challenge dose of morphine (10 mg/kg, i.p.) without AMN082 and/or MMPIP administration and locomotor activity was measured for 60 min. Effect of AMN082 treatment on the expression of sensitization to the locomotor stimulant effect of cocaine and morphine. Influence of MMPIP on the AMN082 effect [3] In order to test the effect of AMN082 on the expression of sensitization to the locomotor stimulant effect of cocaine, separate groups of mice (n = 8–10) were sensitized to cocaine as described above. On the test day (17th day of experiment), mice were treated with AMN082 (1.25, 2.5 or 5.0 mg/kg i.p.) or vehicle 30 min before the cocaine challenge (10 mg/kg, i.p.), and the locomotor activity was measured for 30 min. To determine whether mGluR7s are involved in this AMN082 effect on the expression of cocaine sensitization, the cocaine-sensitized mice, before cocaine challenge (10 mg/kg, i.p.), were pretreated with MMPIP (10 mg/kg, i.p.), 30 min prior to AMN082 (5.0 mg/kg, i.p.) injection. Immediately after cocaine challenge, locomotor activity was measured for 30 min. To evaluate an influence of the mGluR7 agonist on the expression of morphine-induced locomotor sensitization, separate groups of mice (n = 8–10) were sensitized to morphine, as described above. On the test day (20th day of experiment), 30 min prior to the challenge dose of morphine (10 mg/kg, i.p.) mice were treated with AMN082 (1.25, 2.5 or 5.0 mg/kg, i.p.) or vehicle and locomotor activity was recorded for 60 min. To determine whether the mGluR7s are involved in the effect of AMN082 on the expression of morphine sensitization, the morphine sensitized mice, before morphine challenge (10 mg/kg, i.p.), were pretreated with MMPIP (10 mg/kg, i.p.), 30 min before AMN082 (5.0 mg/kg) injection. Immediately after morphine challenge, locomotor activity was measured for 60 min. Effect of AMN082 on the expression of reciprocal locomotor cross-sensitization between cocaine and morphine [3] The induction of cocaine sensitization was performed according to the method described above. Following a period of 4 days without treatment (17th day of experiment), the cocaine-sensitized and saline-treated mice (n = 7–10) were pretreated with AMN082 (2.5 or 5.0 mg/kg, i.p.) or vehicle, 30 min before the challenge with morphine (10 mg/kg, i.p.). Locomotor activity was measured for 30 min, immediately after morphine challenge. The induction of morphine sensitization was performed according to the method described above. Following a period of 7 days without treatment (20th day of experiment) the morphine-sensitized and saline-treated mice (n = 9–10) were pretreated with AMN082 (2.5 or 5.0 mg/kg, i.p.) or vehicle, 30 min before the challenge with cocaine (10 mg/kg, i.p.). Locomotor activity was measured for 60 min, immediately after the cocaine challenge. |
| References |
|
| Additional Infomation |
AMN082 dihydrochloride is a hydrochloride salt prepared by reacting N,N'-bis(diphenylmethyl)ethane-1,2-diamine with two molar equivalents of hydrochloric acid. It possesses the functions of a metabolite glutamate receptor agonist, anti-aging agent, and neuroprotective agent. Its molecular structure contains N,N'-bis(diphenylmethyl)ethane-1,2-diamine (2+).
In this study, we report for the first time the selective mGluR7 agonist AMN082.This compound acts through a previously undescribed site, exhibits oral activity, and selectively modulates the levels of two stress hormones—corticosterone and adrenocorticotropic hormone (ACTH)—in wild-type mice, but not in mGluR7-deficient mice, further confirming the role of mGluR7 in stress physiology. AMN082In the absence of the glutamate site ligand, it induces a complete agonist response comparable to L-AP4. Our data from chimeric receptors strongly suggest the existence of an allosteric agonist site within the transmembrane domain of mGluR7. Further identification of this allosteric site requires the development of suitable radioligands to determine its physicochemical binding properties, such as Kd, Kon, and Koff. However, the absence of AMN082 activity observed in our chimeric receptor data, as well as in other G protein-coupled mGluR proteins expressed in the same host cells used in our mGluR7 function studies, strongly suggests that AMN082's agonist activity derives from its direct interaction with the mGluR7 protein. In particular, this mGluR7-dependent agonist activity was observed in both G protein assays (GTPγS binding) and second messenger assays (cAMP accumulation), significantly reducing the likelihood of AMN082 interacting with components of the intracellular signaling cascade. In conclusion, we have identified a selective mGluR7 agonist, AMN082, which functions through an allosteric site and could serve as a valuable tool for further elucidating the role of mGluR7 in stress-related central nervous system disorders. [1] AMN082 is a selective metabolotropic glutamate receptor mGlu7 agonist, reported to have antidepressant activity. Given that excessive glutamate release is associated with the pathogenesis of depression, this study investigated the effects of N,N'-diphenylmethylethane-1,2-diamine dihydrochloride (AMN082) on glutamate release at nerve endings in the rat cerebral cortex and its possible underlying mechanisms. This study observed that AMN082 inhibited 4-aminopyridine-induced glutamate release, while the metabolotropic glutamate receptor mGlu7 antagonist MMPIP blocked this phenomenon. Furthermore, Western blot analysis and immunocytochemistry confirmed the presence of the presynaptic metabolotropic glutamate receptor mGlu7 protein. Chelating extracellular Ca²⁺ ions and vesicle transporter inhibitors blocked the effect of AMN082 on 4-aminopyridine-induced glutamate release; however, glutamate transporter inhibitors had no effect on the effect of AMN082. AMN082 reduced the 4-aminopyridine-induced increase in intrasynaptic Ca²⁺ concentration but did not alter the synaptosome membrane potential. In the presence of Cav2.2 (N-type) and Cav2.1 (P/Q-type) channel blockers, adenylate cyclase inhibitors, and protein kinase A inhibitors, AMN082 significantly reduced the effect of 4-aminopyridine-induced glutamate release. These results indicate that AMN082 activation of the metabolite glutamate receptor mGlu7 reduces adenylate cyclase/protein kinase A activity, thereby decreasing Ca²⁺ influx through voltage-dependent Ca²⁺ channels and thus reducing induced glutamate release. Furthermore, the clinically effective antidepressant fluoxetine completely blocked the inhibitory effect of AMN082 on glutamate release, suggesting that the two compounds may share the same intracellular mechanism for inhibiting glutamate release from cortical nerve endings. [2] Finally, we propose that the inhibitory effect of AMN082 on 4-aminopyridine-induced glutamate release from cortical nerve endings may be related to the inhibition of the adenylate cyclase/cyclic adenosine monophosphate/protein kinase A pathway. This suggestion is based on the following results: (1) AMN082 inhibited the promoting effect of forskolin on 4-aminopyridine-induced glutamate release; (2) the adenylate cyclase inhibitor MDL12330A and the protein kinase A inhibitor H89 blocked the inhibitory effect of AMN082 on 4-aminopyridine-induced glutamate release. Our findings are consistent with the report by Summa et al. (2013), which showed that AMN082 acts on the metabolite glutamate receptor mGlu7 in hippocampal nerve endings and reduces the production of adenylate cyclase/cyclic adenosine monophosphate, thereby inhibiting the release of γ-aminobutyric acid (GABA). Previous studies have found that the metabotropic glutamate receptor mGlu7 is located presynaptic, and its activation can inhibit glutamate release by reducing the activity of the Gi/o protein-coupled adenylate cyclase/protein kinase A pathway (Millán et al., 2002). Our data show that AMN082 activation of the metabotropic glutamate receptor mGlu7 reduces the activity of adenylate cyclase and protein kinase A, thereby reducing Ca2+ influx through Cav2.2 (N-type) and Cav2.1 (P/Q-type) calcium ion channels, ultimately leading to a reduction in glutamate release induced by cortical nerve endings. In summary, this study confirms that the metabotropic glutamate receptor mGlu7 agonist AMN082 has an inhibitory effect on glutamate release from rat cortical nerve endings. Although the functional role of AMN082 in inhibiting glutamate release explored in this paper is not yet clear, this effect may be a key mechanism for treating brain diseases such as depression, which are associated with excessive glutamate release (Hashimoto et al., 2007). [2] Published data indicate that AMN082 does not affect extracellular glutamate or GABA release in the ventral nucleus accumbens (VP) of cocaine-withdrawing rats (Li et al., 2010), suggesting that glutamate and GABAergic transmission are not involved in the inhibition of cocaine-induced relapse induced by AMN082. This suggests that the mechanisms by which AMN082 antagonizes intravenous cocaine self-administration (via the VP GABAergic mechanism) and the recovery of cocaine-induced drug craving behavior (via the VP glutamate mechanism) may be different. However, studies have shown that VP is involved in the expression of morphine motor sensitization after long-term (3-week) morphine withdrawal rather than short-term (3-day) morphine withdrawal (Mickiewicz et al., 2009). Therefore, more research is needed to fully elucidate the mechanisms by which AMN082 inhibits cocaine and morphine sensitization. This study is the first to demonstrate that the selective mGluR7 allosteric agonist AMN082 can inhibit the development and expression of cocaine and morphine-induced sensitization and cross-sensitization at low doses, and that this low dose does not interfere with the acute hyperkinesis induced by these drugs. These findings suggest that mGluR7 plays an important role in sensitization, as the mGluR7 selective allosteric antagonist MMPIP can block these effects of AMN082. Therefore, our study shows that mGluR7 is involved in the neuroadaptation process associated with cocaine and morphine addiction. Finally, our results indicate that mGluR7 agonists can effectively prevent relapse in cocaine/morphine addicts. [3] |
| Molecular Formula |
C28H29CLN2
|
|---|---|
| Molecular Weight |
428.996266126633
|
| Exact Mass |
464.179
|
| Elemental Analysis |
C, 72.25; H, 6.50; Cl, 15.23; N, 6.02
|
| CAS # |
97075-46-2
|
| Related CAS # |
AMN082 free base;83027-13-8
|
| PubChem CID |
11698390
|
| Appearance |
White to off-white solid powder
|
| LogP |
8.13
|
| Hydrogen Bond Donor Count |
4
|
| Hydrogen Bond Acceptor Count |
2
|
| Rotatable Bond Count |
9
|
| Heavy Atom Count |
32
|
| Complexity |
362
|
| Defined Atom Stereocenter Count |
0
|
| SMILES |
Cl.C1C=CC(C(C2C=CC=CC=2)NCCNC(C2C=CC=CC=2)C2C=CC=CC=2)=CC=1
|
| InChi Key |
YRQCDCNQANSUPB-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C28H28N2.2ClH/c1-5-13-23(14-6-1)27(24-15-7-2-8-16-24)29-21-22-30-28(25-17-9-3-10-18-25)26-19-11-4-12-20-26;;/h1-20,27-30H,21-22H2;2*1H
|
| Chemical Name |
N,N'-dibenzhydrylethane-1,2-diamine;dihydrochloride
|
| Synonyms |
AMN082 DIHYDROCHLORIDE; 97075-46-2; AMN082; AMN 082 dihydrochloride; 83027-13-8; N,N'-Dibenzhydrylethane-1,2-diamine dihydrochloride; AMN 082 (Free base); N1,N2-Dibenzhydrylethane-1,2-diamine dihydrochloride;
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO : ~33.33 mg/mL (~71.61 mM)
|
|---|---|
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples.
Injection Formulations
Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline)(e.g. IP/IV/IM/SC) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). View More
Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] Oral Formulations
Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). View More
Oral Formulation 3: Dissolved in PEG400 (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3310 mL | 11.6550 mL | 23.3100 mL | |
| 5 mM | 0.4662 mL | 2.3310 mL | 4.6620 mL | |
| 10 mM | 0.2331 mL | 1.1655 mL | 2.3310 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved