| Size | Price | Stock | Qty |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
































Purity: ≥98%
| Targets |
125I-SDF-CXCR4 ( IC50 = 13 nM ); HIV-1 (NL4.3 strain) ( IC50 = 1 nM ); HIV-1 (NL4.3 strain) ( IC50 = 9 nM ); HIV-1 (NL4.3 strain) ( IC50 = 3 nM ); HIV-1 (NL4.3 strain) ( IC50 = 26 nM )
CXCR4 (IC50 = 13 nM in a 125I‑SDF‑1 competition binding assay using CEM‑CCRF cells; inhibits SDF‑1 induced Ca²⁺ flux with IC50 = 0.012 μM (12 nM) in CEM‑CCRF cells; inhibits anti‑CXCR4 mAb (clone 12G5) binding with IC50 = 0.013 μM (13 nM) in SUP‑T1 cells)[1] The compound is selective for CXCR4 over other chemokine receptors: CCR1, CCR2b, CCR4, CCR5, CXCR1, CXCR2 (all IC50 > 10 μM in radiolabeled chemokine binding assays).[1] |
|---|---|
| ln Vitro |
AMD-070 is active against X4 strain of HIV-1, HIV-1 IIIb in MT-4 cells, and The IC50 values for AMD-070 are 9-fold higher (0.009 μM vs 0.001 μM) and 8.7-fold higher (0.003 μM vs 0.026 μM) in PBMCs compared to MT-4 cells. AMD-070 has antiviral ability with the IC50 value of 15.5 nM.
Kinase Assay: SUP-T1 T cells are first preincubated with the compounds (with 1 as a control) for 30 min on ice, washed with PBS with 2% FCS, and incubated with PE-conjugated anti-CXCR4 mAb for 30 min on ice. After being washed with PBS, the cell samples are fixed with 1% paraformaldehyde in PBS and analyzed on a FACS Calibur flow cytometery. The dose-dependent inhibitory effects of the compounds on mAb binding are determined using the mean fluorescence intensity values. Cell Assay: he activated cells (PHA-stimulated blasts) are washed three times with PBS, and viral infections are done. HIV-infected or mock-infected PHA-stimulated blasts are cultured in the presence of 25 U/mL of IL-2 and varying concentrations of compounds. Supernatant is collected at day 10, and HIV-1 core antigen in the culture supernatant is analyzed by the p24 viral Ag ELISA kit. Inhibition of HIV-1 replication in MT-4 cells is performed as previously described. Anti-HIV-1 activity and cytotoxicity measurements are carried out in parallel. They are based on the viability of MT-4 cells that has been infected with HIV-1 in the presence of various concentrations of the test compounds. The ICsub>50 is defined as the concentration required to inhibit 50% of the virus-infected cells against viral cytopathicity. In MT‑4 cells infected with HIV‑1 NL4.3 (X4 strain), AMD070 inhibited viral replication with IC50 = 0.002 μM (2 nM) and IC90 = 0.003 μM (3 nM). In PBMCs infected with HIV‑1 NL4.3, IC50 = 0.009 μM (9 nM) and IC90 = 0.026 μM (26 nM). In the presence of 10% human serum, the anti‑HIV‑1 activity of AMD070 against NL4.3 and IIIB strains showed IC50 = 0.016 μM (16 nM) for both. The compound was non‑cytotoxic to MT‑4 cells at concentrations exceeding 23 μM (CC50 > 23 μM).[1] In a CXCR4‑mediated calcium flux assay using CEM‑CCRF cells loaded with Fluo‑4/AM, AMD070 inhibited SDF‑1 (15 nM) induced increase in intracellular calcium with IC50 = 0.012 μM (12 nM) for the (S)-enantiomer (the active enantiomer, which is the drug substance).[1] In a 125I‑SDF‑1 competition binding assay on CEM‑CCRF cells, AMD070 displaced radiolabeled ligand with IC50 = 0.013 μM (13 nM). In an anti‑CXCR4 mAb (clone 12G5) binding assay on SUP‑T1 cells using PE‑conjugated antibody and flow cytometry, AMD070 inhibited antibody binding with IC50 = 0.013 μM (13 nM).[1] Selectivity profiling against other chemokine receptors (CCR1, CCR2b, CCR4, CCR5, CXCR1, CXCR2) using radiolabeled chemokine binding assays (performed at MDS Laboratories) showed that AMD070 had IC50 values > 10 μM for all tested receptors, demonstrating high selectivity for CXCR4.[1] |
| ln Vivo |
AMD-070 shows promising oral bioavailability in rat and dog. The rate of clearance is species dependent with AMD-070 having lower clearance in dog compared to rat.
In BLM-induced pulmonary fibrosis (CD-1 mice, BLM 2 U/kg intratracheal), oral Mavorixafor (AMD-070) HCl at 400 μg/mouse (approx. 20 mg/kg) daily for 21 days did not reduce lung inflammation (5.4% vehicle vs. 14.2% treated by H&E) or fibrosis (3.7% vehicle vs. 10.5% treated by Masson’s Trichrome). However, it significantly improved survival: 90% of AMD070-treated mice survived to day 22 vs. 40% of vehicle-treated mice (P<0.05). [1] In CCl4-induced hepatic fibrosis (C57BL/6 mice, CCl4 1 mL/kg i.p. twice weekly for 4 weeks), intraperitoneal Mavorixafor (AMD-070) HCl at 50 mg/kg (5 days/week, 4 weeks) had no effect on percent liver fibrosis (Picrosirius Red: 3.2% vehicle vs. 3.6% treated), on Acta2 and Col1a1 mRNA levels (by RT-qPCR), or on serum AST levels (elevated similarly in both CCl4 groups). [1] Pharmacodynamic effects: Oral or i.p. administration of Mavorixafor (AMD-070) HCl caused dose-dependent and transient increases in white blood cell counts (primarily lymphocytes), with Tmax at 3 hours, confirming CXCR4 antagonism. [1] |
| Enzyme Assay |
125I‑SDF‑1 competition binding assay: Human CD4⁺CXCR4⁺ CEM‑CCRF cells (5×10⁵ cells) were incubated for 3 h at 4°C in binding buffer (PBS containing 5 mM MgCl₂, 1 mM CaCl₂, 0.25% BSA, pH 7.4) with 100 pM 125I‑SDF‑1α and various concentrations of AMD070 in Millipore Durapore filter plates. Unbound radioligand was removed by washing with 50 mM HEPES, 0.5 M NaCl, pH 7, at 4°C. Bound radioactivity was counted using a 1450 MicroBeta liquid scintillation counter. Data were analyzed by nonlinear regression using GraphPad Prism.[1]
Chemokine receptor selectivity binding assays (CCR1, CCR2b, CCR4, CCR5, CXCR1, CXCR2) were performed at MDS Laboratories (Bothell, WA) using standard radiolabeled chemokine competition methods. IC50 values for AMD070 were all >10 μM.[1] SDF‑1 induced calcium flux assay: CEM‑CCRF cells (10×10⁶ cells/mL) were resuspended in serum‑reduced media (RPMI 1640 with 2% FCS) and loaded with Fluo‑4/AM (1 μM) for 30 min at 37°C. After washing with Hanks balanced salt solution containing 20 mM HEPES, 0.2% BSA, 2.5 mM probenecid (pH 7.4), cells were incubated with AMD070 for 15 min at 37°C. SDF‑1 (15 nM) was added, and changes in intracellular calcium concentration were monitored using a FLEXstation at 525 nm (excitation 494 nm). IC50 values were calculated from dose‑response curves.[1] Anti‑CXCR4 mAb (clone 12G5) binding assay: SUP‑T1 T cells were pre‑incubated with AMD070 for 30 min on ice, washed with PBS containing 2% FCS, then incubated with PE‑conjugated anti‑CXCR4 mAb for 30 min on ice. After washing, cells were fixed with 1% paraformaldehyde in PBS and analyzed by flow cytometry (FACS Calibur). Dose‑dependent inhibition of mAb binding was determined using mean fluorescence intensity values.[1] |
| Cell Assay |
Anti‑HIV‑1 replication assay in MT‑4 cells: The CD4⁺CXCR4⁺ lymphocyte MT‑4 cell line was infected with HIV‑1 NL4.3 (or IIIB) in the presence of various concentrations of AMD070. After several days (typically 5‑7 days), cell viability was measured by the MTT method or similar. The IC50 was defined as the concentration required to inhibit 50% of virus‑induced cytopathicity. The CC50 (50% cytotoxic concentration) was defined as the concentration that reduced viability of mock‑infected cells by 50%.[1]
Anti‑HIV‑1 replication assay in PBMCs: PBMCs from healthy donors were isolated by density gradient centrifugation, stimulated with PHA (1 μg/mL) for 3 days at 37°C, then washed. Activated cells (PHA‑stimulated blasts) were infected with HIV‑1 NL4.3 and cultured in the presence of IL‑2 (25 U/mL) and various concentrations of AMD070. Supernatants were collected on day 10, and HIV‑1 p24 core antigen was measured by ELISA. IC50 and IC90 values were determined.[1] Calcium flux assay details as described under Enzyme Assay (cell‑based functional assay).[1] 12G5 antibody binding assay details as described under Enzyme Assay (cell‑based flow cytometry).[1] |
| Animal Protocol |
2.5 mg/kg, i.v. Rat
Pharmacokinetic studies in rat: Male (or non‑fasted) rats received AMD070 as a solution via intravenous (iv) and oral (po) administration. For iv: dose of 5 mg/kg (or as indicated); for po: dose of 100 mg/kg. Blood samples were collected at various time points (0.5, 1, 2, 3, 6, 24, 48 h post‑dose). Plasma concentrations were determined by protein precipitation followed by UPLC‑MS/MS. Non‑compartmental analysis (WinNonlin) was used to calculate pharmacokinetic parameters. Clearance (Cl) = 3.7 mL/min/kg, volume of distribution (Vdss) = 14.3 L/kg, terminal half‑life (T₁/₂) = approximately 3.5 h, oral bioavailability (F) = 22%.[1] Pharmacokinetic studies in dog: Dogs received AMD070 iv at 2.5 mg/kg and po at 6 mg/kg. Cl = 1.3 mL/min/kg, Vdss = 19.1 L/kg, T₁/₂ = 9.9 h, oral bioavailability F = 80%.[1] Compound formulation: AMD070 was formulated as a solution (the specific vehicle is not detailed in this paper, but typical formulations for oral and iv administration are implied; the compound was used as the hydrochloride salt).[1] |
| ADME/Pharmacokinetics |
Absorption
In adults with WHIM syndrome, after a once-daily dose of 400 mg, the steady-state mean (CV%) Cmax was 3304 (58.6%) ng/mL, and the 0-24 hour AUC (AUC0-24h) was 13970 (58.4%) ng·h/mL. The pharmacokinetics of Mavorixafor are non-linear; the increases in Cmax and AUC0-24h are greater than dose-proportional across the dose range of 50 mg (0.125 times the recommended dose) to 400 mg. In healthy subjects, steady-state plasma concentrations of Mavorixafor are reached approximately after 9 to 12 days following administration of the highest approved recommended dose. At the highest approved recommended dose, the median (range) Tmax of Mavorixafor is 2.8 hours (1.9 to 4 hours). Food decreases Cmax and AUC. Excretion Route In healthy subjects, following a single oral dose of radiolabeled mavolixafor, 74.2% of the administered dose was recovered within a 240-hour collection period, with 61.0% of the radioactive material recovered in feces and 13.2% (3% unchanged) in urine. Volume of Distribution In adults with WHIM syndrome, the volume of distribution of mavolixafor is 768 L. Clearance In healthy subjects, following a single dose of 400 mg mavolixafor, the mean (coefficient of variation) apparent clearance was 62 L/h (40%). Mavolixafor exhibits at least partially nonlinear apparent clearance; however, this nonlinear clearance is not clinically significant at the approved recommended dose. Protein Binding In vitro studies have shown that mavolixafor binds to human plasma proteins >93%. Metabolism/Metabolites Mavolixafor is primarily metabolized via CYP3A4, and secondarily via CYP2D6. Biological Half-Life In healthy subjects, the mean (CV%) terminal half-life after a single dose of 400 mg mavolixafor is 82 hours (34%). Mavorixafor (AMD070) is a small molecule drug with high oral bioavailability; good oral bioavailability was observed in both rats and dogs [1] - In CD-1 mice, after oral administration of 400 μg/mouse of Mavorixafor (AMD070), the drug was detected in lung tissue, and its concentration varied over time. EC₉₀ (44 ng/mL) was used as the reference threshold for pharmacodynamic activity [2] - In C57BL/6 mice, after intraperitoneal injection of Mavorixafor (AMD070), the drug was distributed in plasma, liver, and lung tissue, and its concentration was detected at different time points [2] Rat pharmacokinetics: AMD070 (administered as a solution) showed Cl = 3.7 mL/min/kg, Vdss = 14.3 L/kg, terminal half‑life ≈ 3.5 h (calculated from iv data), oral bioavailability F = 22% (after 100 mg/kg po dose).[1] Dog pharmacokinetics: AMD070 (solution) showed Cl = 1.3 mL/min/kg, Vdss = 19.1 L/kg, terminal half‑life = 9.9 h, oral bioavailability F = 80% (after 6 mg/kg po dose).[1] No other ADME parameters (e.g., metabolism, protein binding, excretion) are reported in this paper.[1] |
| Toxicity/Toxicokinetics |
Mavorixafor (AMD070) showed no cytotoxicity to MT-4 cells and PBMCs at in vitro concentrations exceeding 23 μM[1]
Effects during pregnancy and lactation ◉ Overview of use during lactation There is currently no information regarding the use of mavorixafor during lactation. The manufacturer recommends against breastfeeding during treatment and for three weeks after the last dose. ◉ Effects on breastfed infants No published information was found as of the revision date. ◉ Effects on lactation and breast milk No published information was found as of the revision date. No specific toxicity data for AMD070 are reported in this paper. The paper mentions that a related analogue (15f) was acutely toxic by iv route with an MTD of 1 mg/kg, but that compound was not advanced. For AMD070, the authors state that "2 was selected for safety studies in rat and dog" but no results are provided. No LD50, organ toxicity, or adverse event data are included.[1] |
| References |
PLoS One.2016 Mar 21;11(3):e0151765;J Med Chem.2010 Apr 22;53(8):3376-88.
|
| Additional Infomation |
Mavorixafor is a CXC chemokine receptor 4 (CXCR4) antagonist. It was first approved by the FDA on April 30, 2024, for the treatment of warts, hypogammaglobulinemia, infection, and bone marrow retention (WHIM) syndrome. WHIM syndrome is a inherited immunodeficiency disorder characterized by a decrease in the number of mature neutrophils and lymphocytes. WHIM syndrome is caused by mutations in the CXCR4 gene, leading to overactivation of the CXCR4 signaling pathway. Mavorixafor inhibits CXCR4 activation. Because CXCR4 mutations are also associated with human immunodeficiency virus (HIV), Waldenström macroglobulinemia (WM), B-cell non-Hodgkin lymphoma, and solid tumors including melanoma, Mavorixafor is being investigated in these diseases. Mavorixafor's mechanism of action is as a CXC chemokine receptor 4 antagonist, cytochrome P450 2D6 inhibitor, cytochrome P450 3A4 inhibitor, and P-glycoprotein inhibitor. Mavorixafor is an orally bioavailable CXC chemokine receptor 4 (CXCR4) inhibitor with potential antitumor and immune checkpoint inhibitory activities. After administration, Mavorixafor selectively binds to CXCR4, preventing CXCR4 from binding to its ligand stromal cell-derived factor 1 (SDF-1 or CXCL12). This inhibits receptor activation, leading to reduced proliferation and migration of CXCR4-overexpressing tumor cells. Furthermore, inhibition of CXCR4 prevents the recruitment of regulatory T cells and myeloid-derived suppressor cells (MDSCs) to the tumor microenvironment, thereby eliminating CXCR4-mediated immunosuppression and activating cytotoxic T lymphocyte-mediated immune responses against cancer cells. The G protein-coupled receptor CXCR4 is upregulated in various tumor cell types, inducing the recruitment of immunosuppressive cells to the tumor microenvironment, inhibiting immune surveillance, and promoting tumor angiogenesis and tumor cell proliferation. It is also a co-receptor for HIV entry into T cells. Mavorixafor is a small molecule drug, currently in Phase IV clinical trials (covering all indications), and was first approved in 2024 for the treatment of cancer, with six investigational indications. It is a derivative of AMD3100, a CXCR4 blocker.
AMD070 (also referred to as AMD070, compound 2) is a selective CXCR4 antagonist that was discovered through structure‑activity relationship studies of macrocyclic bicyclam derivatives. It was designed to reduce the overall positive charge at physiological pH compared to earlier compounds (e.g., AMD3100, plerixafor) to improve oral bioavailability. The compound binds to CXCR4, blocking the binding of the natural chemokine SDF‑1 (CXCL12) and inhibiting the entry of X4 (CXCR4‑using) HIV‑1 strains. AMD070 showed high selectivity for CXCR4 over other chemokine receptors (CCR1, CCR2b, CCR4, CCR5, CXCR1, CXCR2). The (S)-enantiomer is the more active isomer, with anti‑HIV‑1 IC50 = 0.002 μM in MT‑4 cells, while the (R)-enantiomer is approximately 50‑fold less potent. The compound was selected for preclinical development and has entered clinical trials for HIV‑1 infection. This paper represents the discovery and optimization leading to AMD070.[1] |
| Molecular Formula |
C21H28CLN5
|
|
|---|---|---|
| Molecular Weight |
385.933523178101
|
|
| Exact Mass |
385.203
|
|
| CAS # |
880549-30-4
|
|
| Related CAS # |
558447-26-0;880549-30-4 (HCl); 2309699-17-8
|
|
| PubChem CID |
71576687
|
|
| Appearance |
Light yellow to brown solid at room temperature
|
|
| LogP |
5.078
|
|
| Hydrogen Bond Donor Count |
3
|
|
| Hydrogen Bond Acceptor Count |
4
|
|
| Rotatable Bond Count |
7
|
|
| Heavy Atom Count |
27
|
|
| Complexity |
431
|
|
| Defined Atom Stereocenter Count |
1
|
|
| SMILES |
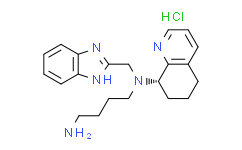
N([C@H]1CCCC2C=CC=NC1=2)(CCCCN)CC1=NC2C=CC=CC=2N1.Cl
|
|
| InChi Key |
DBNMEMJSDAAGNZ-FYZYNONXSA-N
|
|
| InChi Code |
InChI=1S/C21H27N5.ClH/c22-12-3-4-14-26(15-20-24-17-9-1-2-10-18(17)25-20)19-11-5-7-16-8-6-13-23-21(16)19;/h1-2,6,8-10,13,19H,3-5,7,11-12,14-15,22H2,(H,24,25);1H/t19-;/m0./s1
|
|
| Chemical Name |
|
|
| Synonyms |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
|
|||
|---|---|---|---|---|
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples.
Injection Formulations
Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline)(e.g. IP/IV/IM/SC) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). View More
Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] Oral Formulations
Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). View More
Oral Formulation 3: Dissolved in PEG400 (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5911 mL | 12.9557 mL | 25.9114 mL | |
| 5 mM | 0.5182 mL | 2.5911 mL | 5.1823 mL | |
| 10 mM | 0.2591 mL | 1.2956 mL | 2.5911 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved





























 COA
COA



