| Size | Price | Stock | Qty |
|---|---|---|---|
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
































| Targets |
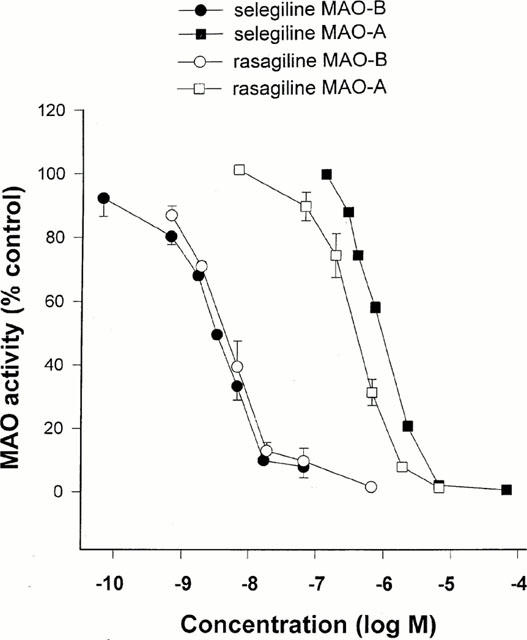
rMAO-B (IC50 = 4.43 nM); rMAO-A (IC50 = 412 nM)
|
|---|---|
| ln Vitro |
Following treatment with dexamethasone (10 µM), the proliferation rate of SH-SY5Y and 1242-MG was considerably boosted by rasagine (0.25 nM; 96 hours) [2].
1. Rasagiline [N-propargyl-1R(+)-aminoindan], was examined for its monoamine oxidase (MAO) A and B inhibitor activities in rats together with its S(-)-enantiomer (TVP 1022) and the racemic compound (AGN-1135) and compared to selegiline (1-deprenyl). The tissues that were studied for MAO inhibition were the brain, liver and small intestine. 2. While rasagiline and AGN1135 are highly potent selective irreversible inhibitors of MAO in vitro and in vivo, the S(-) enantiomer is relatively inactive in the tissues examined. 3. The in vitro IC(50) values for inhibition of rat brain MAO activity by rasagiline are 4.43+/-0.92 nM (type B), and 412+/-123 nM (type A). The ED(50) values for ex vivo inhibition of MAO in the brain and liver by a single dose of rasagiline are 0.1+/-0.01, 0.042+/-0.0045 mg kg(-1) respectively for MAO-B, and 6.48+/-0.81, 2.38+/-0.35 mg kg(-1) respectively for MAO-A. 4. Selective MAO-B inhibition in the liver and brain was maintained on chronic (21 days) oral dosage with ED(50) values of 0.014+/-0.002 and 0.013+/-0.001 mg kg(-1) respectively. 5. The degree of selectivity of rasagiline for inhibition of MAO-B as opposed to MAO-A was similar to that of selegiline. Rasagiline was three to 15 times more potent than selegiline for inhibition of MAO-B in rat brain and liver in vivo on acute and chronic administration, but had similar potency in vitro. 6. These data together with lack of tyramine sympathomimetic potentiation by rasagiline, at selective MAO-B inhibitory dosage, indicate that this inhibitor like selegiline may be a useful agent in the treatment of Parkinson's disease in either symptomatic or L-DOPA adjunct therapy, but lack of amphetamine-like metabolites could present a therapeutic advantage for rasagiline.[1] Stress can affect the brain and lead to depression; however, the molecular pathogenesis is unclear. An association between stress and stress-induced hypersecretion of glucocorticoids occurs during stress. Dexamethasone (a synthetic glucocorticoid steroid) has been reported to induce apoptosis and increase the activity of monoamine oxidase (MAO) (Youdim et al. 1989). MAO is an enzyme for the degradation of aminergic neurotransmitters; dopamine, noradrenaline and serotonin and dietary amines and MAO inhibitors are classical antidepressant drugs. In this study, we have compared the ability of rasagiline (Azilect) and its main metabolite, R-aminoindan with selegiline (Deprenyl) in prevention of dexamethasone-induced brain cell death employing human neuroblastoma SH-SY5Y cells and glioblastoma 1242-MG cells. Dexamethasone reduced cell viability as measured by MTT test, but rasagiline, selegiline, and 1-R-aminoindan could significantly prevent dexamethasone-induced brain cell death. Among three drugs, rasagiline had the highest neuroprotective effect. Furthermore, the inhibitory effects of these drugs on MAO B catalytic activity and on apoptotic DNA damage (TUNEL staining) were examined. Rasagiline exhibited highest inhibition on MAO B enzymatic activity and prevention on DNA damage as compared to selegiline and 1-R-aminoindan. In summary, the greater neuroprotective effect of rasagiline may be associated with the combination of the parent drug and its metabolite 1-R-aminoindan.[2] |
| ln Vivo |
Under a transgenic model of multiple system atrophy, rasagiline is neuroprotective. Treatment with 2.5 mg/kg rasagiline improved motor impairments, according to motor behavior testing [3].
The present study was performed to test the potential of rasagiline as a disease-modifying agent in multiple system atrophy (MSA) using a transgenic mouse model previously described by our group. (PLP)-alpha-synuclein transgenic mice featuring glial cytoplasmic inclusion pathology underwent 3-nitropropionic acid intoxication to model full-blown MSA-like neurodegeneration. Two doses of rasagiline were used (0.8 and 2.5 mg/kg) for a treatment period of 4 weeks. Rasagiline-treated animals were compared to placebo saline-treated mice by evaluation of motor behaviour and neuropathology. Motor behavioural tests including pole test, stride length test and general motor score evaluation showed improvements in motor deficits associated with 2.5 mg/kg rasagiline therapy. Immunohistochemistry and histology showed significant reduction of 3-NP-induced neuronal loss in striatum, substantia nigra pars compacta, cerebellar cortex, pontine nuclei and inferior olives of MSA mice receiving 2.5 mg/kg rasagiline. The results of the study indicate that rasagiline confers neuroprotection in a transgenic mouse model of MSA and may therefore be considered a promising disease-modifying candidate for human MSA.[3] |
| Enzyme Assay |
Determination of MAO inhibitory activity in vitro [1]
The activities of MAO-A and -B were determined by the adapted method of Tipton & Youdim (1983). Rat or human cerebral cortical tissue was homogenized in 0.3 M sucrose (one part tissue to 20 parts sucrose) using a glass-teflon motor-driven homogenizer (brain and liver), or Ultraturrax (gut). The inhibitor under test was added to a suitable dilution of the enzyme preparation in 0.05 M phosphate buffer (pH 7.4) and incubated together with selegiline 0.1 μM (for determination of MAO-A) or clorgyline 0.1 μM (for determination of MAO-B). Incubation was carried on for 60 min at 37°C before addition of labelled substrates (14C-5-hydroxytryptamine creatinine disulphate 100 μM for determination of MAO-A, or 14C-phenylethylamine 10 μM for determination of MAO-B) and incubation continued for 30 or 20 min respectively. The reaction was then stopped by addition of citric acid (2 M). Radioactive metabolites were extracted into toluene/ethyl acetate (1 : 1 v v−1), a solution of 2,5-diphenyloxazole was added to a final concentration of 0.4% (w v−1), and metabolite content estimated by liquid scintillation counting. Activity in presence of drug was expressed as a percentage of that in control samples. [1] The preincubation was carried out in the presence of clorgyline or selegiline because phenylethylamine is also metabolized quite effectively by MAO-A (O'Carroll et al., 1983), leading to inhibition curves for MAO-B, which showed a plateau at about 80% inhibition with selegiline or Rasagiline if MAO-A was not inactivated. For comparison between two inhibitors with potentially different inhibitory effects on MAO-A and MAO-B, therefore, it was thought necessary to employ the system in which opposite enzyme forms are inactivated before assay. MAO B Catalytic Activity Assay [1] SH-SY5Y and 1242-MG cells were grown to confluence, harvested, and washed with phosphate-buffered saline. One hundred micrograms of total proteins were incubated with 10 µM 14C-labeled phenylethylamine in the assay buffer (50 mM sodium phosphate buffer, pH 7.4) at 37°C for 20 min and terminated by the addition of 100 µl of 6 N HCl. The reaction products were then extracted with ethyl acetate/toluene (1:1) and centrifuged at 4°C for 10 min. The organic phase containing the reaction product was extracted, and its radioactivity was obtained by liquid scintillation spectroscopy. |
| Cell Assay |
Cell proliferation assay[2]
Cell Types: Neuroblastoma SH-SY5Y and Glioblastoma 1242-MG Tested Concentrations: 0.25 nM Incubation Duration: 96 hrs (hours) Experimental Results: Increased cell proliferation rate of SH-SY5Y cells treated with dexamethasone About 60%. The cell proliferation rate of 1242-MG cells treated with dexamethasone increased by approximately 35%. Cell Culture and Treatments [2] The SH-SY5Y and 1242-MG cells were seeded into 6-well plates and cultured overnight in medium. Cells were supplemented with charcoal-stripped, steroid-free fetal calf serum for ~6 h. The medium was then replaced with medium treated with 10 µM of dexamethasone, 0.25 nM of Rasagiline, 0.25 nM of selegiline, or 1 µM of 1-R-aminoindan in the presence of charcoal-stripped fetal calf serum. The treatments were performed every other day for 4 days. TUNEL Assay [2] The terminal deoxynucleotidyl transferase (TdT)-mediated dUTP Nick End Labeling (TUNEL) assay was used to assess the extent of apoptosis in treated cells. Briefly, cells were plated on a four-well chamber slide on the day preceding the experiment, and treated with or without 10 µM dexamethasone, 0.25 nM of Rasagiline, 0.25 nM of selegiline, or 1 µM of 1-R-aminoindan for 2 days. Cells were then washed with PBS and fixed using 4% paraformaldehyde in PBS. The slides were again washed with PBS, and fragmented DNA was detected in apoptotic cells by adding fluorescein 12-dUTP to nicked ends of DNA (In Situ Cell Death Detection Kit, Roche). Slides were incubated for 1 h at 37°C in the dark and washed in PBS three times and then visualized with a fluorescent light microscope. Green fluorescence was correlated with DNA fragmentation. Experiments were done in duplicate for three times, and the percentage of TUNEL-positive cells was determined. |
| Animal Protocol |
Animal/Disease Models: (PLP)-α-synuclein transgenic mice over 6 months old [3]
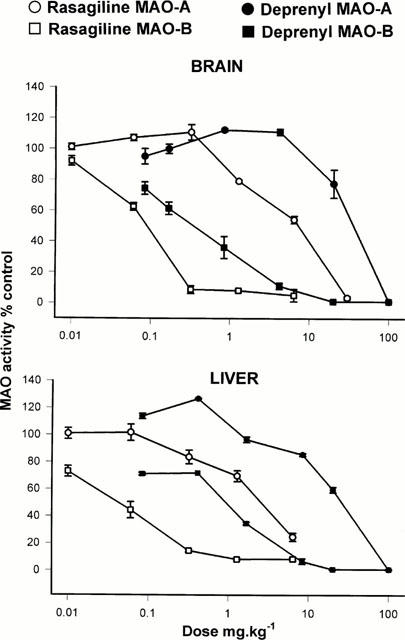
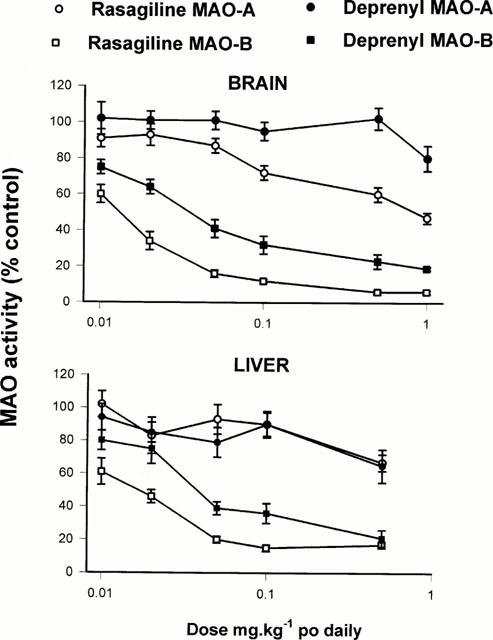
Doses: low dose (0.8 mg/kg bw) and high dose (2.5 mg/kg bw) Route of Administration: every subcutaneous injection once every 24 hrs (hrs (hours)) for 1 time. The total time was 4 weeks (from day 1 to day 28 of the experiment). Experimental Results: Low-dose treatment demonstrated no protective effect in the striatum, with neuronal numbers similar to those in placebo-treated MSA mice. High doses were associated with approximately 15% rescue of DARPP-32-immunoreactive striatal neurons. Low-dose treatment had no effect on nigral neuronal loss, but high-dose treatment completely protected nigral neurons in numbers comparable to healthy controls. Determination of inhibition of MAO activity in vivo [1] In in vivo studies, drugs were administered orally by gavage (p.o.). The animals weighed 250 – 300 g at the time of killing. For estimation of in vivo inhibitory effect, varying doses of the inhibitors were administered to groups of five or six rats for the stated times, the animals were killed by decapitation, tissues removed and frozen at −20°C, and enzyme activity determined subsequently as above. Enzyme activity in drug-treated tissues were expressed as a percentage of that in control tissues. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion
Rasagilan is rapidly absorbed after oral administration. The absolute bioavailability of rasagilan is approximately 36%. Rasagilan undergoes almost complete biotransformation in the liver before excretion. The main elimination pathway is glucuronidation of rasagilan and its metabolites, followed by urinary excretion. After oral administration of 14C-labeled rasagilan, it is primarily eliminated via urine, followed by fecal excretion (62% of the total dose is excreted in urine and 7% in feces within 7 days), with a total recovery rate of 84% within 38 days. Less than 1% of rasagilan is excreted unchanged in the urine. 87 L After oral administration of 14C-labeled rasagilan, it is primarily eliminated via urine, followed by fecal excretion (62% of the total dose is excreted in urine and 7% in feces within 7 days), with a total recovery rate of 84% within 38 days. Less than 1% of rasagiline is excreted unchanged in the urine. Rasagiline is rapidly absorbed; peak plasma concentrations are reached approximately 1 hour after oral administration. The absolute bioavailability of rasagiline is approximately 36%. When taken with a high-fat meal, the peak plasma concentration and area under the plasma concentration-time curve (AUC) of rasagiline decrease by approximately 60% and 20%, respectively; since the AUC is largely unaffected, rasagiline can be taken on an empty stomach or with food. Rasagiline readily crosses the blood-brain barrier. The mean steady-state half-life or terminal half-life of rasagiline is 31 hours and 1.342 hours, respectively; however, there is no correlation between the pharmacokinetic characteristics of rasagiline and its pharmacological effects, as the drug irreversibly inhibits MAO-B, and the recovery of normal enzyme activity depends on the rate of new enzyme synthesis. Rasagiline binds to plasma proteins at a rate of approximately 88-94%, of which 61-63% binds to albumin. In intravenous administration studies in rats and dogs, the volume of distribution (Vd) of rasagiline was several times the total body fluid volume, indicating its extensive tissue distribution. Tissue distribution of 14C-rasagiline was studied in albino and colored rats, showing peak radioactivity in tissues occurring between 0.25 and 0.5 hours. Distribution to the large intestine, bladder, and lacrimal glands was relatively slow, but in the eyes, skin, and arterial walls of colored animals, the duration of drug distribution could reach 24 hours. In vitro animal plasma protein binding was 70% to 90%, and in human plasma protein binding was 88% to 94%. Oral administration of (14)C-rasagiline showed rapid absorption in all species, with peak plasma concentration (Cmax) reached within 2 hours. Absolute bioavailability was estimated at 53% to 69% in rats, 13% to 22% in dogs, and 36% in humans. Toxicokinetic analysis during toxicology studies showed that drug exposure was linear at doses above the level of MOA-B inhibitory pharmacological selectivity, maintaining at approximately 5 mg/kg/day. However, at higher doses, its pharmacokinetics were nonlinear, which may indicate that the elimination of rasagiline and its metabolite aminoindene has reached saturation. In mouse and dog studies, accumulation was observed only at the highest doses (60 and 21 mg/kg/day, respectively). For more complete data on absorption, distribution, and excretion of rasagiline (6 items in total), please visit the HSDB record page. Metabolism/MetabolitesRasagiline undergoes almost complete biotransformation in the liver before excretion. In vitro studies have shown that both metabolic pathways of rasagiline depend on the cytochrome P450 (CYP) system, with CYP 1A2 being the major isoenzyme involved in rasagiline metabolism. After oral administration, rasagiline is extensively metabolized in the liver. In vitro studies have shown that CYP1A2 is the major P450 isoenzyme involved in the metabolic elimination of rasagiline. The major metabolite of rasagiline in human plasma after biotransformation is aminoindene. The main biotransformation pathways of rasagiline in humans include N-dealkylation, indene ring hydroxylation, and phase II N- or O-binding, including N-glucuronidation of the parent drug and its metabolites. In plasma samples from healthy volunteers who have taken rasagiline, the conversion of rasagiline methanesulfonate (R enantiomer) to its S enantiomer was not observed in humans. Rasagiline is not metabolized to amphetamine or methamphetamine. Due to its binding to the MAO site in the intestine before passing through the liver, rasagiline exhibits a significant first-pass metabolic effect. Rasagiline metabolism is rapid and widespread, with similar metabolic characteristics across all tested species. Its main biotransformation pathways are N-dealkylation to aminoindene and hydroxylation to 3-hydroxy-N-propynyl-1-aminoindene. In addition, sulfide or glucuronide conjugation reactions may occur. Microsomal studies have shown that CYP1A2 is the major metabolic isoenzyme, but rasagiline is neither an inducer nor an inhibitor of cytochrome P450. The metabolism of rasagiline under CYP1A2 inhibition, induction, or in the presence of other substrates has been studied clinically. Rasagiline undergoes almost complete biotransformation in the liver before excretion. The metabolism of rasagiline proceeds primarily through two pathways: N-dealkylation and/or hydroxylation, yielding 1-aminoindan (AI), 3-hydroxy-N-propynyl-1-aminoindan (3-OH-PAI), and 3-hydroxy-1-aminoindan (3-OH-AI). In vitro studies have shown that both metabolic pathways of rasagiline depend on the cytochrome P450 (CYP) system, with CYP1A2 being the major isoenzyme involved in rasagiline metabolism. Rasagilan and its metabolites are glucuronidated and subsequently excreted in the urine, which is the primary elimination pathway. Biological Half-Life The mean steady-state half-life of rasagilan is 3 hours, but due to its irreversible inhibition of MAO-B, there is no correlation between its pharmacokinetics and its pharmacological effects. The mean steady-state half-life of rasagilan is 3 hours… After oral administration, in the dose range of 0.5 to 20 mg, the elimination half-life of rasagilan is approximately 0.6 to 2 hours, ranging from 0.3 to 3.5 hours. |
| Toxicity/Toxicokinetics |
Hepatotoxicity
Rasagiline has been reported to cause elevated serum enzymes in a small number of patients taking it long-term, but these abnormalities are usually mild and resolve spontaneously. Rasagiline has not been reported to be associated with cases of acute liver injury, but such cases have been reported with some other nonspecific monoamine oxidase inhibitors. Probability Score: E (Unlikely to cause clinically significant liver injury). Pregnancy and Lactation Effects ◉ Overview of Use During Lactation There are no reports of clinical use of rasagiline during lactation. Rasagiline may lower serum prolactin levels, thus affecting milk production. This may be especially true during the nursing of newborns or premature infants, where alternative medications may be necessary. ◉ Effects on Breastfed Infants As of the revision date, no relevant published information was found. ◉ Effects on Lactation and Breast Milk Animal studies have shown that rasagiline can lower serum prolactin levels. The clinical significance of these findings in lactating women is unclear. For mothers who have established lactation, prolactin levels may not affect their ability to breastfeed. Protein Binding: Plasma protein binding ranges from 88-94%, with an average binding rate of 61-63% to human serum albumin at concentrations of 1-100 ng/ml. Interactions: Antidepressants and selective serotonin reuptake inhibitors (SSRIs): Pharmacological interactions similar to serotonin syndrome may exist (high fever, muscle rigidity, myoclonus, autonomic dysfunction with rapid fluctuations in vital signs and altered mental status, potentially progressing to extreme agitation, delirium, coma, or even death). Concomitant use should generally be avoided. At least 14 days should be elapsed after discontinuing rasagiline before starting SSRIs. Due to the relatively long half-lives of fluoxetine and its main metabolites, the manufacturer of rasagiline recommends at least 5 weeks (longer intervals for high-dose or long-term fluoxetine treatment) after discontinuing fluoxetine before starting rasagiline. CYP1A2 Inhibitors: Pharmacokinetic interactions (elevated plasma rasagiline concentrations) have been observed when used in combination with ciprofloxacin. The dose of rasagiline should be limited in patients taking ciprofloxacin or other CYP1A2 inhibitors. St. John's wort (Hypericum perforatum): Contraindicated with rasagiline. Rasagiline may have potential drug interactions with meperidine (similar to serotonin syndrome), including coma, severe hypertension or hypotension, severe respiratory depression, seizures, malignant hyperthermia, agitation, peripheral vascular failure, and death. Contraindicated with meperidine, methadone, propoxyphene, or tramadol. At least 14 days should be elapsed after discontinuing rasagiline before starting meperidine. For more complete (12 items) data on drug interactions with rasagiline, please visit the HSDB record page. |
| References |
|
| Additional Infomation |
Therapeutic Uses
Rasagiline can be used as initial monotherapy or in combination with levodopa to treat the symptoms of idiopathic Parkinson's disease. Azilec (rasagiline mesylate) is indicated for the treatment of signs and symptoms of idiopathic Parkinson's disease and can be used as initial monotherapy or in combination with levodopa. Azilec has shown efficacy in patients with early-stage Parkinson's disease receiving azilec monotherapy without concurrent dopaminergic therapy. Azilec has shown efficacy as adjunctive therapy in Parkinson's disease patients receiving levodopa. Drug Warnings When used as monotherapy, orthostatic hypotension was reported in approximately 3% of patients receiving 1 mg rasagiline, compared to approximately 5% of patients receiving placebo. In monotherapy trials, orthostatic hypotension did not lead to discontinuation or early withdrawal from the trial in either the rasagiline or placebo group. When rasagiline was used as adjunctive therapy to levodopa, orthostatic hypotension was reported in approximately 6% of patients receiving 0.5 mg rasagiline, approximately 9% of patients receiving 1 mg rasagiline, and approximately 3% of patients receiving placebo. In patients receiving 1 mg/day of rasagiline, one patient (0.7%) discontinued treatment and withdrew from the clinical trial early due to orthostatic hypotension; this did not occur in patients receiving 0.5 mg/day of rasagiline or those receiving placebo. Clinical trial data indicate that orthostatic hypotension most commonly occurs during the first two months of rasagiline treatment and gradually decreases over time. Epidemiological data suggest that patients with Parkinson's disease have a 2 to 4 times higher risk of developing melanoma than the general population; however, it remains unclear whether the observed increased risk is related to the underlying disease or to anti-Parkinson's disease medication. The risk of developing melanoma appears to be higher in patients taking rasagiline than in the general population, but comparable to the risk in patients with Parkinson's disease. Given these findings, patients and clinicians should closely monitor for melanoma development. The manufacturer recommends regular dermatological examinations by a qualified clinician (e.g., a dermatologist); the frequency of these examinations should be determined by the patient's dermatologist. In monotherapy studies, 1.3% of patients receiving 1 mg rasagiline reported hallucinations, compared to 0.7% of patients receiving placebo. In monotherapy trials, 1.3% of patients receiving 1 mg rasagiline discontinued treatment and withdrew from the clinical trial early due to hallucinations, while none of these cases occurred in patients receiving placebo. When used in combination with levodopa, approximately 5% of patients receiving 0.5 mg/day of rasagiline reported hallucinations, approximately 4% of patients receiving 1 mg/day of rasagiline reported hallucinations, compared to approximately 3% of patients receiving placebo. Hallucinations led to discontinuation of treatment and early withdrawal from clinical trials in approximately 1% of patients receiving rasagiline at 0.5 mg/day or 1 mg/day, compared to none in the placebo group. Patients should be informed of the possibility of hallucinations and instructed to immediately inform healthcare professionals if hallucinations occur. The safety and efficacy of rasagiline in children under 18 years of age have not been established. For more complete data on rasagiline (10 total), please visit the HSDB record page. Pharmacodynamics Rasagiline is a propynylamine drug and an irreversible inhibitor of monoamine oxidase (MAO). MAO is a flavin-containing enzyme that regulates the metabolic degradation of catecholamines and serotonin in the central nervous system and peripheral tissues. It exists in two main molecular types, A and B, located in nerve endings throughout the body, the brain, the liver, and the mitochondrial membrane of the intestinal mucosa. MAO-A is mainly found in the gastrointestinal tract and liver, regulating the metabolic degradation of circulating catecholamines and dietary amines. MAO-B is the predominant form in the human brain, responsible for regulating the metabolic degradation of dopamine and phenylethylamine. In in vitro animal studies of brain, liver, and intestinal tissues, rasagiline has been demonstrated as a potent, selective, and irreversible inhibitor of monoamine oxidase type B (MAO-B). At recommended therapeutic doses, rasagiline has also been shown to be a potent and irreversible inhibitor of MAO-B in platelets. Rasagiline selectively inhibits only MAO-B (not MAO-A) in humans, and sensitivity to tyramine during rasagiline treatment at any dose has not been fully characterized, therefore, dietary tyramine and amine intake from the drug cannot be avoided. Drug Indications Azilect is indicated for the treatment of idiopathic Parkinson's disease (PD), as monotherapy (without levodopa) or adjuvant therapy (in combination with levodopa), for patients with end-of-dose fluctuations. This study demonstrates that rasagiline, like selegiline, is an irreversible inhibitor of MAO-B. This conclusion was confirmed through in vitro and in vivo experiments: MAO inhibitory activity in different tissues was measured over a 13-day time interval after oral administration of rasagiline, and the activities of MAO-A and MAO-B were evaluated in vitro. Rasagiline is clearly a highly potent selective MAO-B inhibitor and crosses the blood-brain barrier well, as evidenced by the similarity of inhibition curves in liver and brain tissue. While in vitro experiments showed that rasagiline and selegiline have similar inhibitory efficacy against MAO-B, in vivo studies showed that rasagiline is more potent. This higher efficacy of rasagiline would be even more significant if the dose required to achieve 80% inhibition was measured, rather than 50% of the enzyme inhibition rate. The reason for this is currently unclear, but it may be related to different metabolic rates of the parent compound in vivo or better tissue penetration of rasagiline. Interestingly, preliminary human studies showed that rasagiline is approximately 5 times more potent than selegiline in inhibiting platelet MAO-B (unpublished data). Although rasagiline is more potent than selegiline, its selectivity for inhibiting MAO-A and MAO-B is very similar to that of previously reported selegiline. However, unlike selegiline, the optical isomers of selegiline do not exhibit selectivity for inhibiting MAO-A and MAO-B, while the two optical isomers of AGN 1135 show inhibitory activities for MAO-B and MAO-A that differ by approximately four and two orders of magnitude, respectively. These results are consistent with those of Gotz et al. (1998) in non-human primate (monkey) brain tissue. Gotz et al. administered different doses of rasagiline for seven consecutive days and measured the activity of MAO-A and MAO-B in multiple brain regions, including the caudate nucleus, globus pallidus, cerebral cortex, and hippocampus. Rasagiline was confirmed as a potent and selective inhibitor of MAO-B in the caudate nucleus and globus pallidus, where MAO-B activity is four times that of MAO-A (Gotz et al., 1998). The recovery of MAO-A and MAO-B activity after in vivo inhibition is related to the synthesis of enzymatic apolipoproteins, and the recovery varies among different tissues (liver, intestine, and brain). The small intestine shows the fastest recovery of MAO-B activity, while the brain shows the slowest. This difference in enzyme activity recovery in rat tissues after rasagiline treatment is not uncommon, as similar recovery has been reported after inhibition with selegiline and clogiline (Neff and Goridis, 1972; Della Corte and Tipton, 1980). In fact, the half-life of MAO-B recovery after selegiline treatment has been reported to exceed 30 days in the primate (monkey and human) brain (Fowler et al., 1994) and to be 13 days in the rat brain (Neff and Goridis, 1972; Della Corte and Tipton, 1980). In summary, this study shows that rasagiline is a potent and irreversible MAO-B inhibitor, with a potency 3-15 times greater than selegiline in rats, and similar selectivity for the inhibition of MAO-B and MAO-A. Due to its purer pharmacological properties, lack of amphetamine-like characteristics, and the fact that its metabolite is aminoindene rather than 1-methylamphetamine, and its recent reports of neuroprotective and anti-apoptotic properties (Finberg et al., 1998; Huang et al., 1999; Youdim et al., 1999), we can conclude that this drug may have better activity than selegiline in the treatment of Parkinson's disease. [1] We are the first to report that rasagiline, selegiline, and 1-R-aminoindene can significantly inhibit dexamethasone-induced brain cell death, including neuroblastoma and glioblastoma cells. Among these three compounds, rasagiline has the strongest neuroprotective effect, which is superior to selegiline and 1-R-aminoindene. Rasagiline (Azilect) and selegiline (1-deprazole or Emsam) are irreversible inhibitors of MAO B. The stronger neuroprotective effect of rasagiline may be partly attributed to its parent compound and its major metabolite, 1-R-aminoindene. Furthermore, the inhibitory effects of these drugs on MAO B catalytic activity and apoptotic DNA fragment damage (observed by TUNEL staining) were investigated. Rasagiline showed the strongest inhibition of MAO B enzyme activity (Youdim et al., 2001a), and also exhibited the strongest inhibition of apoptosis compared to selegiline and 1-R-aminoindene. The mechanism by which rasagiline and selegiline exert their anti-apoptotic effects can be summarized as follows: they upregulate the anti-apoptotic proteins Bcl-2 and Bcl-X1, and downregulate the pro-apoptotic proteins Bad, Bax, PARP, and caspase 3 (see reviews by Youdim et al., 2005a and Youdim et al., 2006 for details). Since Bcl-2 and caspase 3 are key factors in preventing or mediating mitochondrial-related apoptosis (Lakhani et al., 2006), it is suggested that MAO inhibitors may protect cells from apoptosis by maintaining mitochondrial homeostasis (Malorni et al., 1998). Furthermore, structure-activity relationship studies of rasagiline indicate that the propargylamine moiety is responsible for this effect, as propargylamine has little or no monoamine oxidase (MAO) inhibitory activity but possesses a similar neuroprotective mechanism and potency (Bar-Am et al., 2005). In addition, both rasagiline and propargylamine activate neuroprotective protein kinases C (PKCα and PKCε) while downregulating pro-apoptotic PKCδ and γ. Inhibition of PKC with GF109203X blocks their neuroprotective activity (Weinreb et al., 2005; Youdim et al., 2005a). The mechanism of action of aminoindane has been fully elucidated. This study used a cytotoxic model of human neuroblastoma SKN-SH cells to evaluate the neuroprotective effect of 1-R-aminoindene under high-density culture-induced neuronal death and 6-hydroxydopamine stimulation. The results showed that 1-R-aminoindene (0.1–1 µM) significantly reduced the level of the apoptosis-associated phosphorylated protein H2A.X (Ser139), decreased the cleavage of caspase 9 and caspase 3, and increased the levels of anti-apoptotic proteins Bcl-2 and Bcl-xl. The protein kinase C (PKC) inhibitor GF109203X blocked this neuroprotective effect, indicating that PKC is involved in aminoindene-induced cell survival. Aminoindene significantly increased pPKC (pan-) levels, particularly the level of the pro-survival PKC subtype PKCε (Bar-Am et al., 2007). In summary, the neuroprotective activity of rasagiline and its major metabolite 1-R-aminoindene observed in this study and previous studies may be related to the results of the recent prospective clinical study ADAGIO for Parkinson's disease patients. The ADAGIO study showed that early treatment with rasagiline can provide benefits that cannot be obtained with later treatment. This is the first prospective, large-scale, randomized, double-blind trial to provide evidence that the drug may delay the progression of Parkinson's disease through neuroprotective effects (Hughes, 2008) [2]. |
| Molecular Formula |
C12H13N
|
|---|---|
| Molecular Weight |
171.2383
|
| Exact Mass |
171.104
|
| Elemental Analysis |
C, 84.17; H, 7.65; N, 8.18
|
| CAS # |
136236-51-6
|
| Related CAS # |
Rasagiline mesylate;161735-79-1;Rasagiline-13C3 mesylate racemic;1216757-55-9
|
| PubChem CID |
3052776
|
| Appearance |
Light yellow to brown <38°C powder,>41°C liquid
|
| Density |
1.1±0.1 g/cm3
|
| Boiling Point |
305.5±30.0 °C at 760 mmHg
|
| Melting Point |
35-41°C
|
| Flash Point |
146.8±20.0 °C
|
| Vapour Pressure |
0.0±0.6 mmHg at 25°C
|
| Index of Refraction |
1.577
|
| LogP |
2.27
|
| Hydrogen Bond Donor Count |
1
|
| Hydrogen Bond Acceptor Count |
1
|
| Rotatable Bond Count |
2
|
| Heavy Atom Count |
13
|
| Complexity |
212
|
| Defined Atom Stereocenter Count |
1
|
| SMILES |
N([H])(C([H])([H])C#C[H])[C@@]1([H])C2=C([H])C([H])=C([H])C([H])=C2C([H])([H])C1([H])[H]
|
| InChi Key |
RUOKEQAAGRXIBM-GFCCVEGCSA-N
|
| InChi Code |
InChI=1S/C12H13N/c1-2-9-13-12-8-7-10-5-3-4-6-11(10)12/h1,3-6,12-13H,7-9H2/t12-/m1/s1
|
| Chemical Name |
(1R)-N-prop-2-ynyl-2,3-dihydro-1H-inden-1-amine
|
| Synonyms |
rasagiline; 136236-51-6; (R)-N-(2-Propynyl)-2,3-dihydroinden-1-amine; Azilect; (R)-N-2-Propynyl-1-indanamine; (1R)-N-(prop-2-yn-1-yl)-2,3-dihydro-1H-inden-1-amine; 1-Indanamine, N-2-propynyl-, (R)-; TV-1030;
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO : ~100 mg/mL (~583.98 mM)
H2O : ≥ 5.88 mg/mL (~34.34 mM) |
|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (14.60 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (14.60 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. View More
Solubility in Formulation 3: ≥ 2.5 mg/mL (14.60 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 5.8398 mL | 29.1988 mL | 58.3976 mL | |
| 5 mM | 1.1680 mL | 5.8398 mL | 11.6795 mL | |
| 10 mM | 0.5840 mL | 2.9199 mL | 5.8398 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03727139 | Completed Has Results | Drug: Rasagiline | Parkinson's Disease | Takeda | November 1, 2018 | |
| NCT01879748 | Completed | Drug: Rasagiline Drug: Placebo |
Parkinson's Disease | Teva Branded Pharmaceutical Products R&D, Inc. |
June 2013 | Phase 1 |
| NCT01032486 | Completed | Drug: Rasagiline mesylate | Parkinson's Disease | Teva Branded Pharmaceutical Products R&D, Inc. |
December 2009 | |
| NCT00203164 | Completed | Drug: rasagiline mesylate | Parkinson's Disease | Teva Branded Pharmaceutical Products R&D, Inc. |
May 2002 | Phase 3 |
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved