| Size | Price | Stock | Qty |
|---|---|---|---|
| 50mg |
|
||
| 100mg |
|
||
| 250mg | |||
| Other Sizes |
Purity: ≥98%
QX-314 Br is a novel intracellular blocker of NaV channels1-4 and a membrane impermeable quaternary analogue of lidocaine, a blocker of voltage-activated Na+ channels. QX 314 bromide also inhibits calcium currents in hippocampal CA1 pyramidal neurons.
| Targets |
Na+/Sodium channel
|
|---|---|
| ln Vitro |
On the transient potential vanilloid subtype 1 channel (TRPV1), QX-314 chloride exhibits biphasic effects in vitro [2]. Concentrations of QX-314 chloride ranging from 1 to 60 mM immediately and potentiometrically activate TRPV1[2]. In hippocampus CA1 pyramidal neurons, intracellular calcium currents are inhibited by QX-314 chloride (10 mM), resulting in low-threshold (T-type) Ca2+ currents that average less than 45% of control amplitude [2].
QX-314 activated TRPV1 channels at 10, 30, and 60 mM (0.4 ± 0.1%, 3.5 ± 1.3%, and 21.5 ± 6.9% of normalized peak activation, respectively; mean ± SEM; n = 12) but not TRPV4 channels (P < 0.001). Activation by QX-314 was blocked by the TRPV1 antagonist, capsazepine (100 μM). QX-314 (60 mM) activation and blockade by capsazepine was also demonstrated in Ca²⁺ imaging studies on TRPV1-expressing tsA201 cells. At subactivating concentrations (less than 1 mM), QX-314 potently inhibited capsaicin-evoked TRPV1 currents with an IC₅₀ of 8.0 ± 0.6 μM. Conclusions: The results of this study show that the quaternary lidocaine derivative QX-314 exerts biphasic effects on TRPV1 channels, inhibiting capsaicin-evoked TRPV1 currents at lower (micromolar) concentrations and activating TRPV1 channels at higher (millimolar) concentrations. These findings provide novel insights into the interactions between QX-314 and TRPV1 and may provide an explanation for the irritant properties of intrathecal QX-314 in mice in vivo. [1] 1. The effects of intracellular QX-314 on Ca2+ currents were examined in CA1 pyramidal cells acutely isolated from rat hippocampus. In neurons dialyzed with 10 mM QX-314 (bromide salt), the amplitude of the high-threshold Ca2+ current was on average 20% of that in control cells and the current-voltage relationships (I-Vs) were shifted in the positive voltage direction. 2. The positive shift in the I-Vs was due to the presence of intracellular Br-, because it was reproduced by 10 mM NaBr and was not present when the chloride salt of QX-314 was used. 3. Low-threshold (T-type) Ca2+ currents, at test voltages of -50 and -40 mV, were on average < 45% of control amplitude in cells containing 10 mM QX-314 (chloride salt) and < 10% of control amplitude in cells with 10 mM QX-314 (bromide salt). 4. In neurons dialyzed with 1 mM QX-314, high-threshold Ca2+ currents were still significantly different from control and Na+ currents were not completely blocked. 5. The proportions of high-threshold Ca2+ current blocked by omega-conotoxin GVIA, omega-agatoxin IVA, and nimodipine were similar in cells dialyzed with 10 mM QX-314 and control cells, indicating that the drug does not selectively inhibit any of the Ca2+ channel subtypes distinguished by these antagonists [4]. Transient receptor potential vanilloid subtype 1 (TRPV1) channels are subjected to biphasic effects by QX-314 bromide in vitro [1]. TRPV1 is immediately activated by QX-314 bromide (1–60 mM) in a concentration-dependent manner [1]. Cell death and blackening of the oocyte membrane are caused by QX-314 bromide (≥ 30 mM) [1]. In hippocampal CA1 pyramidal neurons, QX-314 bromide suppresses intracellular calcium currents, resulting in low-threshold (T-type) Ca2+ currents that average less than 10% of the control amplitude [4]. Because QX-314 bromide contains intracellular Br-, it causes a shift in the current-voltage relationship (IV) toward a positive voltage [4]. |
| ln Vivo |
QX-314 bromide (1.6 mg/kg; ic) eliminates reactions to painful mechanical and thermal stimuli when combined with capsaicin without causing impairments to motor or tactile function [2].
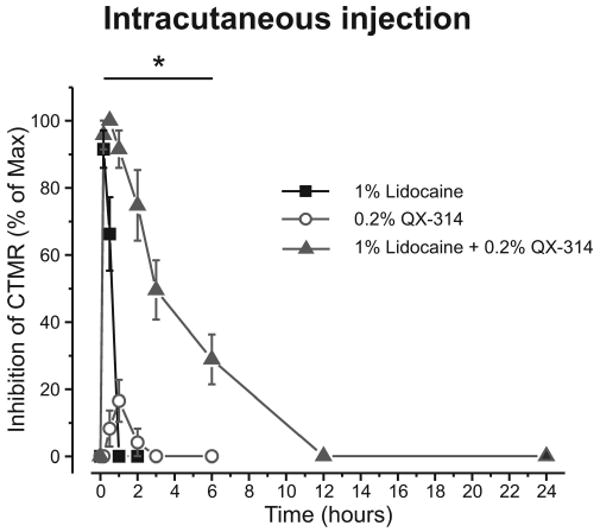
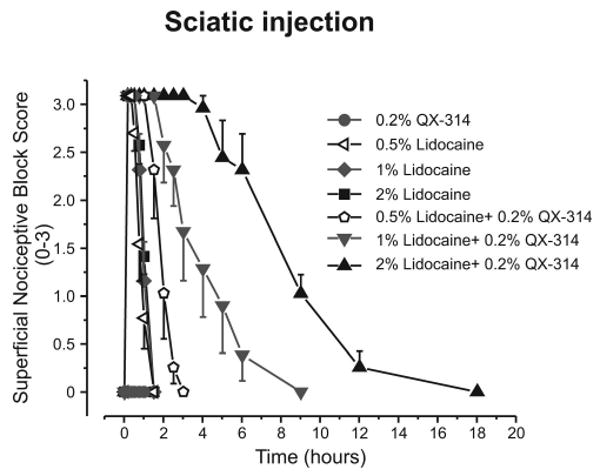
Here, we investigated the pain response of Tnxb-/- mice using pharmacological silencing of A-fibers with co-injection of N-(2,6-Dimethylphenylcarbamoylmethyl) triethylammonium bromide (QX-314), a membrane-impermeable lidocaine analog, plus flagellin, a toll-like receptor 5 (TLR5) ligand. Intraplantar co-injection of QX-314 and flagellin significantly increased the paw withdrawal threshold to transcutaneous sine wave stimuli at frequencies of 250 Hz (Aδ fiber responses) and 2000 Hz (Aβ fiber responses), but not 5 Hz (C fiber responses) in wild-type mice. The QX-314 plus flagellin-induced silencing of Aδ- and Aβ-fibers was also observed in Tnxb-/- mice. Co-injection of QX-314 and flagellin significantly inhibited the mechanical allodynia and neuronal activation of the spinal dorsal horn in Tnxb-/- mice. Interestingly, QX-314 alone inhibited the mechanical allodynia in Tnxb-/- mice, and it increased the paw withdrawal threshold to stimuli at frequencies of 250 Hz and 2000 Hz in Tnxb-/- mice, but not in wild-type mice. The inhibition of mechanical allodynia induced by QX-314 alone was blocked by intraplantar injection of a TLR5 antagonist TH1020 in Tnxb-/- mice. These results suggest that mechanical allodynia due to TNX deficiency is caused by the hypersensitivity of Aδ- and Aβ-fibers, and it is induced by constitutive activation of TLR5.[3] Coapplication of 0.2% QX-314 with lidocaine prolonged the nociceptive block relative to lidocaine alone, an effect attenuated in TRPV1 knockout mice. The 0.2% QX-314 alone had no effect when injected intraplantary or perineurally, and it produced only weak short-lasting inhibition of the cutaneous trunci muscle reflex. Perisciatic nerve injection of lidocaine with QX-314 produced a differential nociceptive block much longer than the transient motor block, lasting 2 h (for 1% lidocaine) to 9 h (2% lidocaine). Triple application of lidocaine, QX-314, and capsaicin further increased the duration of the differential block. Conclusions: Coapplication of lidocaine and its quaternary derivative QX-314 produces a long-lasting, predominantly nociceptor-selective block, likely by facilitating QX-314 entry through TRPV1 channels. Delivery of QX-314 into nociceptors by using lidocaine instead of capsaicin produces sustained regional analgesia without nocifensive behavior.[2] |
| Enzyme Assay |
The authors conducted an in vitro laboratory study in which they expressed TRPV1 and TRPV4 channels in Xenopus laevis oocytes and recorded cation currents with the two-electrode voltage clamp method. They used confocal microscopy for Ca²⁺ imaging in TRPV1 transient transfected tsA201 cells. Drugs were bath-applied by gravity perfusion. Statistical analyses were performed using Student t test, ANOVA, and post tests as appropriate (P < 0.05).
Results: QX-314 activated TRPV1 channels at 10, 30, and 60 mM (0.4 ± 0.1%, 3.5 ± 1.3%, and 21.5 ± 6.9% of normalized peak activation, respectively; mean ± SEM; n = 12) but not TRPV4 channels (P < 0.001). Activation by QX-314 was blocked by the TRPV1 antagonist, capsazepine (100 μM). QX-314 (60 mM) activation and blockade by capsazepine was also demonstrated in Ca²⁺ imaging studies on TRPV1-expressing tsA201 cells. At subactivating concentrations (less than 1 mM), QX-314 potently inhibited capsaicin-evoked TRPV1 currents with an IC₅₀ of 8.0 ± 0.6 μM.[1]
|
| Cell Assay |
1. The effects of intracellular QX-314 on Ca2+ currents were examined in CA1 pyramidal cells acutely isolated from rat hippocampus. In neurons dialyzed with 10 mM QX-314 (bromide salt), the amplitude of the high-threshold Ca2+ current was on average 20% of that in control cells and the current-voltage relationships (I-Vs) were shifted in the positive voltage direction. 2. The positive shift in the I-Vs was due to the presence of intracellular Br-, because it was reproduced by 10 mM NaBr and was not present when the chloride salt of QX-314 was used. 3. Low-threshold (T-type) Ca2+ currents, at test voltages of -50 and -40 mV, were on average < 45% of control amplitude in cells containing 10 mM QX-314 (chloride salt) and < 10% of control amplitude in cells with 10 mM QX-314 (bromide salt). 4. In neurons dialyzed with 1 mM QX-314, high-threshold Ca2+ currents were still significantly different from control and Na+ currents were not completely blocked. 5. The proportions of high-threshold Ca2+ current blocked by omega-conotoxin GVIA, omega-agatoxin IVA, and nimodipine were similar in cells dialyzed with 10 mM QX-314 and control cells, indicating that the drug does not selectively inhibit any of the Ca2+ channel subtypes distinguished by these antagonists.[2]
|
| Animal Protocol |
Animal/Disease Models: Male SD (SD (Sprague-Dawley)) rat (250-290 g) [2]
Doses: 1.6 mg/kg Route of Administration: intradermal injection Experimental Results: Response to noxious mechanical and thermal stimuli was eliminated when co-treated with capsaicin , without motor or tactile deficits. QX-314 was dissolved in a saline solution. Capsazepine (1 mg/mL) was dissolved in saline containing 5% dimethyl sulfoxide and 5% Tween 20. Mice were injected subcutaneously into the plantar surface of the hind paw with 20 μL of QX-314 (30 mM), QX-314 (30 mM) plus flagellin (0.5 μg in 20 μL), QX-314 (30 mM) plus capsaicin (10 μg in 20 μL), and TH1020 (10 nmol, 4.5 μg), using 27-gauge stainless steel needle attached to a microsyringe. For pre-administration of TH1020 or capsazepine, mice were subcutaneously injected with 10 μL of TH1020 (10 nmol in 10 μL), capsazepine (10 μg in 10 μL), or vehicle, and then 10 μL of QX-314 (60 mM) was administrated 30 min after administration. All drug administration experiments were performed by investigators blinded to the drugs.[3] QX-314 chloride, QX-314 bromide, and N -methyl-d-glucamine chloride were directly dissolved in the extracellular solution (see Electrophysiology). The pH values of all solutions were adjusted to pH 7.4 with N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid] (HEPES) and NaOH. All control and test solutions were applied with an automated fast-switching multibarrel perfusion system [1]. Intraplantar Injections [2] Intraplantar injections of QX-314 (lidocaine N-ethyl bromide) (0.2%, 10 μl, 1.6 mg/kg; Sigma, St. Louis, MO) and/or lidocaine-HCl (1%, 10 μl, 8 mg/kg) into the left hind paw (n = 6 for each group) were made; mechanical and thermal sensitivities were determined by using von Frey hairs,3 and radiant heat of 54°C4 focused on an 8 × 8-mm plantar skin area, respectively, at the times indicated. Intracutaneous Injections [2] Rats were briefly anesthetized by inhalation of 1–2% sevoflurane. Drug solutions were injected subcutaneously via the shaved dorsal thoracolumbar region. The injection with 0.3 ml volume resulted in a circular wheal, which was then marked with ink. Groups of eight rats were injected with each test solution: lidocaine (1%), QX-314 (0.2%), and lidocaine mixed with QX-314. Sciatic Nerve Injections [2] Rats were lightly anesthetized by inhalation of sevoflurane, and the landmarks (greater trochanter and ischial tuberosity) of the left hind limb were localized. Groups of eight rats were injected with 0.2 ml of each test solution: lidocaine (0.5%, 4 mg/kg; 1%, 8 mg/kg; 2%, 16 mg/kg), QX-314 (0.2%), and lidocaine mixed with QX-314 (0.5% lidocaine + 0.2% QX-314, 1% lidocaine + 0.2% QX-314, 2% lidocaine + 0.2% QX-314). The drug was injected in immediate proximity to the sciatic nerve with a 27-gauge hypodermic needle attached to a tuberculin syringe as described,5 and the rat was observed for the development of sciatic nerve block, indicated by complete paralysis of the hind limb. For some experiments, coinjection of lidocaine (1% and 2%) and 0.2% QX-314 was followed by injection of 0.05% of capsaicin (10 min apart). The right hind limb was used as a control. Motor function and nocifensive reactions were evaluated immediately before inhalation of sevoflurane at, 10, 20, 30, 45, 60, 90, 120, 150, and 180 min, and then at 4, 5, 6, 9, 12, 18, and 24 h or until complete recovery. Trpv1 Knockouts [2] TRPV1 knockout mice on a C57BL/6 background, and male wild-type (WT) C57BL/6 mice were housed at a temperature of 23 ± 2°C with a 12-h light-dark cycle (light on 08:00 to 20:00), and fed food and water ad libitum. The animals were allowed to habituate to the housing facilities for 1 week before the experiments. Lidocaine and QX-314 were used. Intraplantar Injections and Behavior Testing [2] Mechanical threshold was assessed by measuring foot withdrawal thresholds in response to mechanical stimuli to the right hind paw. The 50% withdrawal threshold was determined using the up-down method with a set of von Frey filaments (0.02–6 g). Mice were placed in a plastic cage with a wire mesh bottom and were allowed at least 30 min for behavioral accommodation. After pretest thresholds were determined by two sets of experiments, 10 μl of 5% lidocaine, 5% lidocaine with 0.2% QX-314, 0.2% QX-314, or normal saline was injected into the plantar aspect of the right hindpaw plantar. Then, thresholds were measured at 10, 20, 30, 60, 90, 120, 180, 240, and 300 min or until complete recovery. All tests were performed blinded. QX-314 was dissolved in a saline solution. Capsazepine (1 mg/mL) was dissolved in saline containing 5% dimethyl sulfoxide and 5% Tween 20. Mice were injected subcutaneously into the plantar surface of the hind paw with 20 μL of QX-314 (30 mM), QX-314 (30 mM) plus flagellin (0.5 μg in 20 μL), QX-314 (30 mM) plus capsaicin (10 μg in 20 μL), and TH1020 (10 nmol, 4.5 μg), using 27-gauge stainless steel needle attached to a microsyringe. For pre-administration of TH1020 or capsazepine, mice were subcutaneously injected with 10 μL of TH1020 (10 nmol in 10 μL), capsazepine (10 μg in 10 μL), or vehicle, and then 10 μL of QX-314 (60 mM) was administrated 30 min after administration. All drug administration experiments were performed by investigators blinded to the drugs.[3] |
| Toxicity/Toxicokinetics |
The intraperitoneal LD50 in mice was 25 mg/kg. (Acta Pharmaceutica Suecica., 2(213), 1965)
|
| References |
|
| Additional Infomation |
Background: Transient receptor potential vanillic acid subfamily member 1 (TRPV1) channels are important integrators of nociceptive stimuli and are significantly expressed in nociceptive neurons. The experimental local anesthetic QX-314, a quaternary ammonium (i.e., permanently charged) lidocaine derivative, has recently been shown to interact with and penetrate these channels, thereby producing nociceptive and sensory blocking effects in animals. However, the specific interaction between QX-314 and TRPV1 channels is poorly understood. Therefore, the authors investigated the mechanism by which QX-314 acts on TRPV1 channels. Activation by QX-314 (60 mM) and blockade of TRPV1 channels by capsaicin were also confirmed in Ca²⁺ imaging studies on TRPV1-expressing tsA201 cells. At subactivational concentrations (below 1 mM), QX-314 effectively inhibited capsaicin-induced TRPV1 currents, with an IC₅₀ of 8.0 ± 0.6 μM. Conclusion: The results of this study indicate that the quaternary lidocaine derivative QX-314 has a biphasic effect on the TRPV1 channel, inhibiting capsaicin-induced TRPV1 currents at low (µmol) concentrations and activating the TRPV1 channel at higher (millimol) concentrations. These findings provide new insights into the interaction between QX-314 and TRPV1 and may explain the stimulatory properties of intrathecal QX-314 in mice. [1] In summary, in this work, we demonstrated, through experiments conducted on Xenopus oocytes and tsA201 cells, that the quaternary lidocaine derivative QX-314 acts as a biphasic regulator of expressed human TRPV1 channels. At low µmol concentrations, QX-314 inhibits TRPV1 activation in the presence of capsaicin; while at millimol concentrations, which are more relevant to clinical regional anesthesia and nerve blocks, QX-314 is a lidocaine-like TRPV1 channel agonist. 28Our results indicate that QX-314 has a dual effect on specific molecular targets, which not only describes a novel property of what we consider to be quaternary ammonium local anesthetics, but may also provide a new molecular mechanism explanation for adverse reactions caused by local anesthetics in regional anesthesia. [1]When the permanently charged lidocaine derivative QX-314 is co-administered with capsaicin, a transient receptor potential vanillic acid receptor 1 (TRPV1) channel agonist, QX-314 enters the nociceptor, thereby producing nociceptive selective local anesthesia. However, capsaicin-induced pain limits its clinical application until QX-314-mediated blockade is established. Since lidocaine can also activate the TRPV1 channel, the authors tested whether lidocaine could replace capsaicin to deliver QX-314 into the nociceptor through the TRPV1 channel, thereby producing selective analgesia. Methods: Lidocaine (0.5% [17.5 mM], 1% [35 mM] and 2% [70 mM]), QX-314 (0.2% [5.8 mM]), and combinations thereof were subcutaneously injected near the sciatic nerve in rats and mice. Mechanical and thermal responses and motor blockade were measured. [2] In summary, we found that in rodents, the combination of lidocaine and QX-314 produced a sustained regional analgesia; the duration of analgesia was even longer when QX-314 was combined with lidocaine and capsaicin in triplicate. Although lidocaine itself is transient and nonselective, making this method less nociceptive than using capsaicin as a TRPV1 agonist, it may be clinically superior because it avoids the transient but intense initial pain response mediated by TRPV1 channel activation while still producing a sustained regional analgesia that lasts much longer than motor blockade. Furthermore, the combination of lidocaine and QX-314 may be clinically ideal, as it induces relatively short motor blockade followed by more durable regional analgesia, thereby achieving initial fixation at the surgical site and maintaining analgesia after the recovery of motor function. [2] In summary, we demonstrated that mechanical hyperalgesia caused by TNX deficiency is mediated by A-fiber hypersensitivity by inhibiting sensory fibers through pharmacological methods and by using QX-314 in combination with flagellin. In addition, in Tnxb−/− mice, QX-314 alone inhibited A-fiber response and mechanical hyperalgesia by constitutive activation of TLR5. Our findings will contribute to the treatment of neurological complications in patients with EDS. [3]
|
| Molecular Formula |
C16H27BRN2O
|
|---|---|
| Molecular Weight |
343.30238366127
|
| Exact Mass |
342.131
|
| Elemental Analysis |
C, 55.98; H, 7.93; Br, 23.27; N, 8.16; O, 4.66
|
| CAS # |
24003-58-5
|
| Related CAS # |
QX-314 chloride;5369-03-9
|
| PubChem CID |
9884487
|
| Appearance |
White to off-white solid powder
|
| LogP |
0.195
|
| Hydrogen Bond Donor Count |
1
|
| Hydrogen Bond Acceptor Count |
2
|
| Rotatable Bond Count |
6
|
| Heavy Atom Count |
20
|
| Complexity |
268
|
| Defined Atom Stereocenter Count |
0
|
| SMILES |
[Br-].O=C(C[N+](CC)(CC)CC)NC1C(C)=CC=CC=1C
|
| InChi Key |
DLHMKHREUTXMCH-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C16H26N2O.BrH/c1-6-18(7-2,8-3)12-15(19)17-16-13(4)10-9-11-14(16)5/h9-11H,6-8,12H2,1-5H31H
|
| Chemical Name |
N-(2,6-Dimethylphenylcarbamoylmethyl)triethylammonium bromide
|
| Synonyms |
QX-314 Br; QX-314Br; QX314 Br; 24003-58-5; N-(2,6-Dimethylphenylcarbamoylmethyl)triethylammonium bromide; QX-314 BROMIDE; 3OYF1S84EN; UNII-3OYF1S84EN; DTXSID001018052; Ethanaminium, 2-((2,6-dimethylphenyl)amino)-N,N,N-triethyl-2-oxo-, bromide (1:1); Ethanaminium, 2-[(2,6-dimethylphenyl)amino]-N,N,N-triethyl-2-oxo-, bromide (1:1); QX 314; QX314.
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
H2O : ~100 mg/mL (~291.29 mM)
DMSO : ~50 mg/mL (~145.65 mM) |
|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (6.06 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (6.06 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. View More
Solubility in Formulation 3: ≥ 2.08 mg/mL (6.06 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. Solubility in Formulation 4: 50 mg/mL (145.65 mM) in PBS (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.9129 mL | 14.5645 mL | 29.1290 mL | |
| 5 mM | 0.5826 mL | 2.9129 mL | 5.8258 mL | |
| 10 mM | 0.2913 mL | 1.4565 mL | 2.9129 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved





























































 COA
COA