| Size | Price | Stock | Qty |
|---|---|---|---|
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g | |||
| Other Sizes |
































Purity: ≥98%
Olanzapine (formerly LY-170052, LY 170052, Zyprexa, Zolafren), a thienobenzodiazepine analog, is an approved atypical antipsychotic drug with high affinity for 5-HT2 serotonin and D2 dopamine receptor. It functions as a 5-HT2 antagonist of dopamine and serotonin. According to binding studies, olanzapine exhibited a nanomolar affinity for dopaminergic, serotonergic, alpha 1-adrenergic, and muscarinic receptors, and interacted with key receptors of interest in schizophrenia. The U.S. FDA has approved it for the treatment of bipolar disorder and schizophrenia. Olanzapine and quetiapine share structural similarities.
| Targets |
5-HT2A Receptor ( Ki = 4 nM ); 5-HT1 Receptor ( Ki = 7 nM ); 5-HT6 Receptor ( Ki = 5 nM ); 5-HT2C Receptor ( Ki = 11 nM ); 5-HT3 Receptor ( Ki = 57 nM ); Adrenergic α1 Receptor ( Ki = 19 nM ); Muscarinic M1-5 Receptor ( Ki = 1.9-25 nM ); Dopamine Receptor; Mitophagy; Apoptosis
|
||
|---|---|---|---|
| ln Vitro |
In vitro activity: Olanzapine has a nanomolar affinity for dopaminergic, serotonergic, alpha 1-adrenergic, and muscarinic receptors, which allows it to interact with important receptors implicated in schizophrenia. Similar to clozapine, olanzapine exhibits a receptor profile that is largely nonselective at dopamine receptor subtypes, with selectivity for mesolimbic and mesocortical dopamine tracts over striatal dopamine tracts (electrophysiology; Fos). [1]
Olanzapine has little or no effect at other receptors, enzymes, or key proteins in neuronal function. Olanzapine has a receptor profile that is similar to that of clozapine: it is relatively nonselective at dopamine receptor subtypes and it shows selectivity for mesolimbic and mesocortical over striatal dopamine tracts (electrophysiology; Fos). Conclusion: The binding and functional profile of olanzapine (1) is similar to that of clozapine, (2) indicates that olanzapine is an atypical antipsychotic drug, and (3) is consistent with clinical efficacy. If olanzapine also proves to be safe, then it will have high potential to become a more ideal antipsychotic drug [1]. |
||
| ln Vivo |
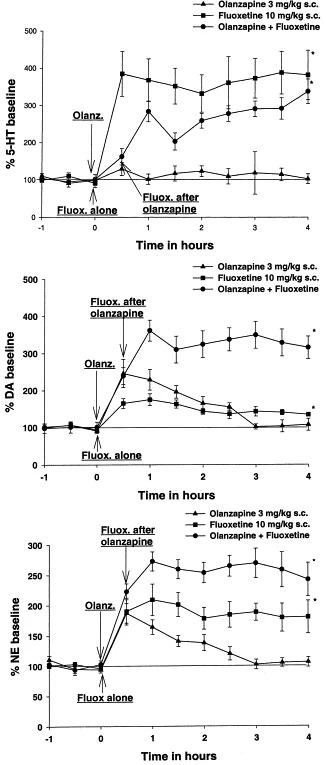
Olanzapine is a weaker antagonist at alpha-adrenergic and muscarinic receptors, but a strong antagonist at DA receptors (DOPAC levels; pergolide-stimulated increases in plasma corticosterone) and 5-HT receptors (quipazine-stimulated increases in corticosterone).[1] In the rat prefrontal cortex, Olanzapine plus fluoxetine together result in strong, prolonged increases in extracellular levels of norepinephrine ([NE](ex)) and dopamine ([DA](ex)) up to 361% and 272% of the baseline, respectively. These increases are noticeably higher than those caused by either medication alone.[2] In rat prefrontal cortex, nucleus accumbens, and striatum, olanzapine at 0.5 mg/kg, 3 mg/kg, and 10 mg/kg (s.c.) dose-dependently raises the extracellular dopamine (DA) and norepinephrine (NE) levels. Olanzapine also raises the concentrations of 3-methoxytyramine, a released DA metabolite, in tissues and extracellular levels of DOPAC, another DA metabolite.[3] Fresh brain weights on average and left cerebrum volumes and fresh weights in macaque monkeys are reduced by 8–11% when olanzapine is administered. [4] Obesity is significantly elevated when taking olanzapine; elevated visceral and subcutaneous adipose stores are reflected in elevated total body fat. Hepatic insulin resistance is brought on by olanzapine.[5]
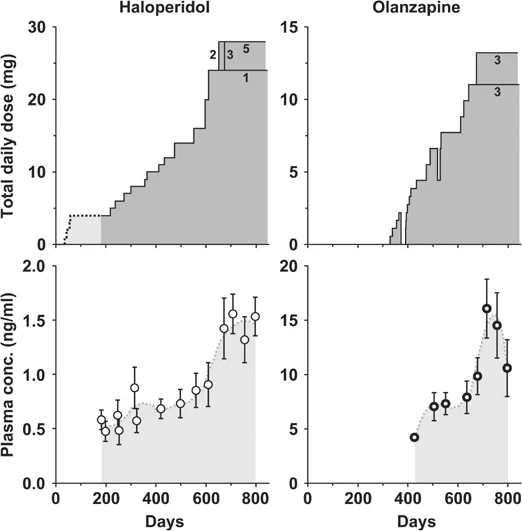
In vivo Olanzapine is a potent antagonist at DA receptors (DOPAC levels; pergolide-stimulated increases in plasma corticosterone) and 5-HT receptors (quipazine-stimulated increases in corticosterone), but is weaker at alpha-adrenergic and muscarinic receptors.[1] To understand the mechanism of the clinical efficacy of Olanzapine and fluoxetine combination therapy for treatment-resistant depression (TRD), we studied the effects of olanzapine and other antipsychotics in combination with the selective serotonin uptake inhibitors fluoxetine or sertraline on neurotransmitter release in rat prefrontal cortex (PFC) using microdialysis. The combination of olanzapine and fluoxetine produced robust, sustained increases of extracellular levels of dopamine ([DA](ex)) and norepinephrine ([NE](ex)) up to 361 +/- 28% and 272 +/- 16% of the baseline, respectively, which were significantly greater than either drug alone. This combination produced a slightly smaller increase of serotonin ([5-HT](ex)) than fluoxetine alone. The combination of clozapine or risperidone with fluoxetine produced less robust and persistent increases of [DA](ex) and [NE](ex). The combination of haloperidol or MDL 100907 with fluoxetine did not increase the monoamines more than fluoxetine alone. Olanzapine plus sertraline combination increased only [DA](ex). Therefore, the large, sustained increase of [DA](ex), [NE](ex), and [5-HT](ex) in PFC after olanzapine-fluoxetine treatment was unique and may contribute to the profound antidepressive effect of the olanzapine and fluoxetine therapy in TRD. [3] It is unclear to what degree antipsychotic therapy confounds longitudinal imaging studies and post-mortem studies of subjects with schizophrenia. To investigate this problem, we developed a non-human primate model of chronic antipsychotic exposure. Three groups of six macaque monkeys each were exposed to oral haloperidol, Olanzapine or sham for a 17-27 month period. The resulting plasma drug levels were comparable to those seen in subjects with schizophrenia treated with these medications. After the exposure, we observed an 8-11% reduction in mean fresh brain weights as well as left cerebrum fresh weights and volumes in both drug-treated groups compared to sham animals. The differences were observed across all major brain regions (frontal, parietal, temporal, occipital, and cerebellum), but appeared most robust in the frontal and parietal regions. Stereological analysis of the parietal region using Cavalieri's principle revealed similar volume reductions in both gray and white matter. In addition, we assessed the subsequent tissue shrinkage due to standard histological processing and found no evidence of differential shrinkage due to drug exposure. However, we observed a pronounced general shrinkage effect of approximately 20% and a highly significant variation in shrinkage across brain regions. In conclusion, chronic exposure of non-human primates to antipsychotics was associated with reduced brain volume. Antipsychotic medication may confound post-mortem studies and longitudinal imaging studies of subjects with schizophrenia that depend upon volumetric measures.[4] Atypical antipsychotics have been linked to weight gain, hyperglycemia, and diabetes. We examined the effects of atypical antipsychotics Olanzapine (OLZ) and risperidone (RIS) versus placebo on adiposity, insulin sensitivity (S(I)), and pancreatic beta-cell compensation. Dogs were fed ad libitum and given OLZ (15 mg/day; n = 10), RIS (5 mg/day; n = 10), or gelatin capsules (n = 6) for 4-6 weeks. OLZ resulted in substantial increases in adiposity: increased total body fat (+91 +/- 20%; P = 0.000001) reflecting marked increases in subcutaneous (+106 +/- 24%; P = 0.0001) and visceral (+84 +/- 22%; P = 0.000001) adipose stores. Changes in adiposity with RIS were not different from that observed in the placebo group (P > 0.33). Only OLZ resulted in marked hepatic insulin resistance (hepatic S(I) [pre- versus postdrug]: 6.05 +/- 0.98 vs. 1.53 +/- 0.93 dl . min(-1) . kg(-1)/[microU/ml], respectively; P = 0.009). beta-Cell sensitivity failed to upregulate during OLZ (pre-drug: 1.24 +/- 0.15, post-drug: 1.07 +/- 0.25 microU . ml(-1)/[mg/dl]; P = 0.6). OLZ-induced beta-cell dysfunction was further demonstrated when beta-cell compensation was compared with a group of animals with adiposity and insulin resistance induced by moderate fat feeding alone (+8% of calories from fat; n = 6). These results may explain the diabetogenic effects of atypical antipsychotics and suggest that beta-cell compensation is under neural control [5]. |
||
| Enzyme Assay |
Method: We evaluated olanzapine interactions with neuronal receptors using standard assays of radioreceptor binding in vitro and well-established in vivo (functional) assays.
Results: Binding studies showed that olanzapine interacts with key receptors of interest in schizophrenia, having a nanomolar affinity for dopaminergic, serotonergic, alpha 1-adrenergic, and muscarinic receptors [1]. |
||
| Animal Protocol |
|
||
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion
Olanzapine exhibits linear pharmacokinetics, reaching steady-state plasma concentrations approximately one week after daily administration. At normal doses, steady-state plasma concentrations of olanzapine appear not to exceed 150 ng/ml, with an AUC of 333 ng/h/ml. Food does not affect olanzapine absorption. The pharmacokinetic profile of olanzapine is a peak plasma concentration of 156.9 ng/ml approximately 6 hours after oral administration. Olanzapine is primarily excreted via metabolism; therefore, only 7% of the drug is excreted unchanged. Olanzapine is mainly excreted in the urine (approximately 53% of the excreted dose) and secondarily in feces (approximately 30%). The reported volume of distribution for olanzapine is 1000 liters, indicating its extensive distribution in the body. The mean clearance of olanzapine is 29.4 L/h, but some studies report an apparent clearance of 25 L/h. This study examined the excretion of olanzapine in the breast milk of five lactating women with postpartum psychosis. Nine pairs of plasma and breast milk samples were collected, and olanzapine concentrations were determined using high-performance liquid chromatography (HPLC). Single-point breast milk/plasma ratios were calculated, ranging from 0.2 to 0.84, with a mean of 0.46. The median relative dose to the infants was 1.6% (range 0–2.5%) of the weight-adjusted maternal dose. No significant adverse reactions were observed in infants during the study period due to exposure to these doses of olanzapine. As with other antipsychotics, this study indicates that olanzapine enters breast milk. … Olanzapine is distributed in breast milk. The manufacturer notes that in a study of healthy lactating women, the estimated mean steady-state olanzapine dose to infants was approximately 1.8% of the maternal olanzapine dose. In another study assessing olanzapine exposure in infants of seven lactating women (who received 5-20 mg olanzapine daily for 19-395 days), the median and maximum relative doses observed in infants were 1% and 1.2%, respectively. Olanzapine was not detected in the plasma of breastfed infants, and no adverse events possibly associated with olanzapine exposure were reported in infants in this study. Furthermore, the peak drug concentration in breast milk occurred, on average, 5.2 hours later than the corresponding peak drug concentration in maternal plasma. In one case report, a woman estimated an infant's relative dose of approximately 4% based on measurements of drug concentrations in serum and breast milk after 4 and 10 days of olanzapine treatment (estimated to have reached steady state). Intramuscular olanzapine is rapidly absorbed, with peak plasma drug concentrations occurring within 15 to 45 minutes. A pharmacokinetic study in healthy volunteers showed that the peak plasma drug concentration produced by intramuscular injection of 5 mg olanzapine was on average approximately five times that produced by oral administration of 5 mg olanzapine. The area under the curve (AUC) after intramuscular administration was similar to that after oral administration of the same dose. The half-life after intramuscular administration was similar to that after oral administration. Olanzapine pharmacokinetics is linear within the clinical dose range. Olanzapine is widely distributed throughout the body, with a volume of distribution of approximately 1000 liters. In the concentration range of 7 to 1100 ng/mL, it binds to plasma proteins in 93% of its volume, primarily albumin and α1-acid glycoprotein. Olanzapine is well absorbed after oral administration, reaching peak plasma concentration in approximately 6 hours. Olanzapine is primarily eliminated via first-pass metabolism, with approximately 40% of the dose metabolized before entering systemic circulation. Food does not affect the rate or extent of olanzapine absorption. Pharmacokinetic studies have shown that olanzapine tablets and orally disintegrating tablets are bioequivalent. Olanzapine is primarily metabolized in the liver, accounting for approximately 40% of the administered dose, mainly through glucuronidase and the cytochrome P450 system. In the cytochrome P450 system, the major metabolic enzymes are CYP1A2 and CYP2D6. As part of phase I metabolism, the major circulating metabolites of olanzapine (accounting for approximately 50-60% of this phase) are 10-N-glucuronide and 4'-N-demethylolanzapine, which are clinically inactive and catalyzed by CYP1A2. On the other hand, CYP2D6 catalyzes the formation of 2-OH olanzapine, while flavin-containing monooxygenase (FMO3) is responsible for the formation of N-oxide olanzapine. In phase II metabolism of olanzapine, UGT1A4 is a key enzyme that generates the direct-bound form of olanzapine. The metabolic profile after intramuscular administration is similar in nature to that after oral administration. Direct glucuronidation and cytochrome P450 (CYP)-mediated oxidation are the main metabolic pathways of olanzapine. In vitro studies have shown that CYP1A2 and 2D6, as well as the flavin-containing monooxygenase system, are involved in the oxidation of olanzapine. CYP2D6-mediated oxidation appears to be a minor metabolic pathway in vivo, as olanzapine clearance was not reduced in subjects lacking this enzyme. Following a single oral dose of 14C-labeled olanzapine, 7% of the dose was recovered in urine, indicating active olanzapine metabolism. Approximately 57% and 30% of the dose were recovered in urine and feces, respectively. In plasma, olanzapine accounted for only 12% of the total radioactive AUC, indicating significant exposure to metabolites. After multiple doses, the major circulating metabolites were 10-N-glucuronide (44% of olanzapine at steady-state concentration) and 4'-N-desmethylolanzapine (31% of olanzapine at steady-state concentration). Neither metabolite showed pharmacological activity at the observed concentrations. Known metabolites of olanzapine include olanzapine N-oxide, 2-hydroxymethylolanzapine, N-desmethylolanzapine, and 7-hydroxyolanzapine. Biological Half-Life The half-life of olanzapine is 21 to 54 hours, with a mean half-life of 30 hours. The half-life range is 21 to 54 hours (5th to 95th percentiles; mean 30 hours). |
||
| Toxicity/Toxicokinetics |
Toxicity Overview
Olanzapine alone has low toxicity. However, there are case reports of olanzapine poisoning when taken in combination with other drugs at high doses. For example, one case report described a patient who experienced extreme hypotension, circulatory failure, respiratory depression, and coma after an overdose of olanzapine (560 mg), propranolol (6.4 g), and amlodipine (280 mg). According to product labels and post-marketing reports, olanzapine poisoning is characterized by the following: A serum olanzapine concentration >0.1 mg/L indicates poisoning, and a serum concentration >1 mg/L can be fatal. Clinical Manifestations Agitation Dysarthria Tachycardia and Hypotension Extrapyramidal Symptoms Sedation Miosis Aspiration Delirium Respiratory Depression Coma Seizures Ventricular Arrhythmias Management There is no specific antidote for olanzapine. In case of acute overdose, an airway should be established and maintained, ensuring adequate oxygenation and ventilation, including endotracheal intubation. Furthermore, clinicians should consider the possibility of multiple drug interactions. In addition, gastric lavage (if the patient is comatose, after endotracheal intubation) and administration of activated charcoal and laxatives should be considered. Administration of activated charcoal (1 gram) can reduce the Cmax and AUC of oral olanzapine by approximately 60%. Because olanzapine blood concentrations typically reach peak levels approximately 6 hours after administration, activated charcoal may be an effective treatment for olanzapine overdose. Overdose may cause confusion, seizures, or head and neck dystonia, which may lead to aspiration risk and may induce vomiting. Therefore, cardiovascular monitoring, including continuous electrocardiographic monitoring, should be initiated immediately to detect any possible arrhythmias. Hypotension and circulatory failure should be treated appropriately, such as with intravenous fluids and sympathomimetic drugs. Do not use adrenaline, dopamine, or other sympathomimetic drugs with beta-agonist activity, as beta-receptor stimulation may worsen hypotension in the presence of olanzapine-induced alpha-receptor blockade. Close medical monitoring and surveillance should continue until the patient recovers. The antipsychotic activity of olanzapine may derive from its antagonistic effects on D2 receptors in the mesolimbic pathway and 5-HT2A receptors in the frontal cortex. D2 receptor antagonism can alleviate positive symptoms of schizophrenia, while 5-HT2A receptor antagonism can alleviate negative symptoms. Olanzapine is an antagonist of dopamine receptors types 1, 2, and 4, 5-HT receptors types 2A and 2C, muscarinic receptors types 1 to 5, α1 receptors, and histamine H1 receptors. Olanzapine's antipsychotic effect stems from its antagonistic effect on dopamine and 5-HT2 receptors, with higher activity against 5-HT2 receptors than against dopamine type 2 receptors. This may explain its lack of extrapyramidal side effects. Olanzapine does not appear to block dopamine within the tuberous-infundibular bundle, which explains its lower incidence of hyperprolactinemia compared to typical antipsychotics or risperidone. Olanzapine also has antagonistic effects on muscarinic receptors, H1 receptors, and α1 receptors. Hepatotoxicity: It has been reported that 10% to 50% of patients taking olanzapine long-term develop abnormal liver function. These abnormalities are usually mild, asymptomatic, and transient, and are reversible with continued use. In addition, patients taking olanzapine have been reported to have significantly elevated serum transaminase levels and clinically significant hepatitis with jaundice. Among atypical antipsychotics, olanzapine is most closely associated with clinically significant liver injury, with an estimated incidence of 1/1200. Olanzapine-induced liver injury usually occurs within 1 to 4 weeks after initiation of treatment or reaching the optimal daily dose. However, there are reports of liver injury occurring up to one year after initiation of treatment. The most common pattern of elevated serum enzymes is mixed (cases), but hepatocellular or cholestatic patterns can also occur. There have been reports of death due to olanzapine-induced liver injury, but most cases recover rapidly upon discontinuation of olanzapine. Allergic reactions (rash, fever, eosinophilia) and autoimmune markers are uncommon. Cases with a long latency period and significant weight gain may suggest non-alcoholic fatty liver disease rather than olanzapine hepatotoxicity. Drug Interactions The manufacturer notes that olanzapine clearance in smokers is approximately 40% higher than in non-smokers. Therefore, plasma olanzapine concentrations are typically lower in smokers than in non-smokers when taking the drug. One patient treated with olanzapine reported extrapyramidal adverse reactions after reducing smoking, while another patient treated with olanzapine reported increased delusions, hostility, and aggressive behavior after a significant increase in smoking (i.e., from 12 cigarettes per day to 80). Although the exact mechanism of this interaction is not fully understood, studies suggest that smoking components (especially CYP1A2) inducing CYP isoenzymes may be partly responsible for the lower plasma olanzapine concentrations in smokers compared to non-smokers. While the manufacturer states that routine dose adjustments are not recommended for smokers taking olanzapine, some clinicians suggest monitoring smoking levels in patients receiving olanzapine and considering dose adjustments for patients experiencing reduced or increased smoking, poor response, or dose-related adverse reactions. In addition, monitoring plasma olanzapine concentrations may be helpful for smokers and patients with other factors associated with significant alterations in olanzapine metabolism (e.g., elderly patients, women, or those taking fluvoxamine concurrently). Concomitant administration of activated charcoal (1 gram) can reduce peak plasma concentration and AUC by approximately 60% after a single dose of 7.5 mg olanzapine. Since peak plasma concentration is typically reached approximately 6 hours after oral administration, activated charcoal may be helpful in treating olanzapine toxicity. Olanzapine treatment may enhance the effects of certain antihypertensive drugs when used concurrently. Furthermore, dopamine, epinephrine, and/or other sympathomimetic drugs with β-receptor agonist activity should be avoided when treating olanzapine-induced hypotension, as such stimulation may exacerbate hypotension in the presence of α-receptor blockade caused by olanzapine. In a pharmacokinetic study, concomitant administration of a single dose of alcohol did not significantly alter the steady-state pharmacokinetics of olanzapine (up to 10 mg daily). However, concomitant use of olanzapine with alcohol enhances olanzapine-related orthostatic hypotension. Therefore, the manufacturer recommends avoiding alcohol consumption during olanzapine treatment. For more complete data on olanzapine drug interactions (11 items in total), please visit the HSDB record page. |
||
| References | |||
| Additional Infomation |
Therapeutic Uses
Antiemetic, Antipsychotic, Serotonin Reuptake Inhibitor Oral olanzapine is indicated for the treatment of schizophrenia. Its efficacy has been demonstrated in three clinical trials in adult patients with schizophrenia: two 6-week trials and one maintenance therapy trial. Its efficacy was also demonstrated in a 6-week trial in adolescent patients with schizophrenia (13-17 years of age). /US Product Label Includes/ Oral olanzapine in combination with fluoxetine is indicated for the treatment of depressive episodes associated with bipolar I disorder, based on clinical studies in adult patients. /US Product Label Includes/ Oral olanzapine is indicated for the treatment of manic or mixed episodes associated with bipolar I disorder, as adjunctive therapy to lithium or valproate. Its efficacy has been demonstrated in two 6-week clinical trials in adults. Long-term efficacy of adjunctive therapy has not been systematically evaluated in controlled trials. /US Product Label Includes/ For more complete data on the therapeutic uses of olanzapine (7 types), please visit the HSDB record page. Drug Warning /Black Box Warning/ Warning: Increased Mortality in Patients with Alzheimer's Disease-Related Psychosis. Patients with Alzheimer's disease-related psychosis receiving antipsychotic medication have an increased risk of death. An analysis of 17 placebo-controlled trials (mean duration 10 weeks) showed that the risk of death was 1.6 to 1.7 times higher in patients receiving medication compared to those receiving placebo. These trials primarily involved patients taking atypical antipsychotics. In typical 10-week controlled trials, the mortality rate was approximately 4.5% in the medication group and approximately 2.6% in the placebo group. Although the causes of death varied, most deaths appeared to be related to cardiovascular diseases (e.g., heart failure, sudden death) or infectious diseases (e.g., pneumonia). Observational studies have shown that, similar to atypical antipsychotics, treatment with conventional antipsychotics may increase mortality. The extent to which the increased mortality observed in observational studies is attributable to the antipsychotics themselves, rather than certain characteristics of the patients, is currently unclear. Olanzapine is not approved for the treatment of patients with dementia-related psychosis. It has been reported that taking antipsychotic medications, including olanzapine, can lead to a potentially fatal syndrome, sometimes referred to as neuroleptic malignant syndrome (NMS). Clinical manifestations of NMS include high fever, muscle rigidity, altered mental status, and autonomic dysfunction (irregular pulse or blood pressure, tachycardia, excessive sweating, and arrhythmias). Other signs may include elevated creatine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure. Diagnostic evaluation of this syndrome is complex. At the time of diagnosis, it is essential to rule out cases where clinical manifestations are accompanied by serious medical conditions (e.g., pneumonia, systemic infection) and untreated or inadequately treated extrapyramidal symptoms (EPS). Other important considerations in differential diagnosis include central anticholinergic toxicity, heatstroke, drug fever, and primary central nervous system disorders. Treatment of NMS should include: 1) immediate discontinuation of antipsychotic medications and other unnecessary medications; 2) intensive symptomatic treatment and medical monitoring; and 3) treatment of any accompanying serious medical problems requiring specific treatment. There is currently no consensus on specific drug treatment regimens for NMS. If a patient requires antipsychotic medication after NMS recovery, the possibility of reintroducing medication should be carefully considered. Due to reports of NMS relapse, patients should be closely monitored. Schizophrenia and type I bipolar disorder are inherently suicidal; therefore, high-risk patients should be closely monitored while on medication. Olanzapine prescriptions should use the lowest possible dose, adhering to good patient management principles, to minimize the risk of overdose. Like other atypical antipsychotics, olanzapine is less likely to cause certain extrapyramidal adverse reactions (such as dystonia). Results from controlled clinical trials indicate that extrapyramidal reactions associated with olanzapine treatment are dose-related. In controlled clinical trials, approximately 4% of patients receiving oral olanzapine and approximately 1% of patients receiving intramuscular olanzapine reported tremor; the incidence of tremor appears to be dose-related. In addition, approximately 3% of patients receiving oral olanzapine experienced akathisia, compared to less than 1% of those receiving intramuscular olanzapine; in short-term controlled clinical trials, approximately 3% of patients receiving oral olanzapine experienced increased muscle tone. For more complete data on olanzapine warnings (45 in total), please visit the HSDB record page. Pharmacodynamics: Olanzapine has been reported to produce positive effects by acting on D2 receptors, such as reducing hallucinations, delusions, speech disturbances, thought disturbances, and behavioral disturbances. On the other hand, olanzapine's action on serotonin 2A receptors can prevent symptoms such as anhedonia, emotional blunting, poverty of speech, decreased willpower, and poor concentration. Based on its specific mechanism of action, olanzapine has a higher affinity for dopamine D2 receptors compared to other dopamine receptor subtypes. This characteristic significantly reduces the incidence of side effects. Olanzapine, initially used to treat schizophrenia and adult bipolar disorder, as well as acute manic or mixed episodes of bipolar disorder in adolescents, demonstrated significant efficacy in clinical trials. Studies have shown that olanzapine's action on dopamine and serotonin receptors can alleviate chemotherapy-induced nausea and vomiting, as these receptors are believed to be involved in this process. Several clinical trials have been conducted targeting this effect, showing that olanzapine significantly improves overall control of nausea and vomiting. In a high-level study of olanzapine for this disease, 84% of patients observed complete remission during the delayed phase, and over 80% of patients maintained control of vomiting during the delayed phase. Background: Classic (typical) antipsychotics are widely used clinically, but some patients do not respond completely to treatment, while others experience no improvement in negative symptoms and cognitive deficits. Furthermore, these drugs often cause severe motor disorders. Clozapine, an atypical antipsychotic, appears to correct many of these deficits, but its incidence is high and it can lead to fatal agranulocytosis. Therefore, we attempted to develop a more effective and safer next-generation antipsychotic prototype. Our strategy was to develop a compound that not only exhibits activity in behavioral tests predicting antipsychotic effects but also possesses the rich and multifaceted receptor pharmacology of clozapine, while avoiding its side effects. To this end, Eli Lilly developed olanzapine. This article elucidates the in vitro and in vivo receptor pharmacology of olanzapine. Methods: We evaluated the interaction of olanzapine with neuronal receptors using standard in vitro radioactive receptor binding assays and well-established in vivo (functional) assays. Results: Combined studies showed that olanzapine interacts with key receptors in schizophrenia, exhibiting nanomolar affinity for dopaminergic, serotonergic, α1-adrenergic, and muscarinic receptors. In vivo experiments demonstrated that olanzapine is a potent antagonist of dopamine receptors (DOPAC levels; elevated plasma corticosterone levels after pergolitide stimulation) and serotonin receptors (elevated corticosterone levels after quiperazine stimulation), but with weaker antagonism towards α1-adrenergic and muscarinic receptors. Olanzapine has little effect on other receptors, enzymes, or key proteins in neuronal function. Olanzapine has a similar receptor profile to clozapine: it is relatively less selective for dopamine receptor subtypes and more selective for mesolimbic and mesocortical dopamine pathways than striatal dopamine pathways (electrophysiology; Fos). Conclusion: The binding and functional characteristics of olanzapine (1) are similar to those of clozapine, (2) suggesting that olanzapine is an atypical antipsychotic, and (3) consistent with clinical efficacy. If olanzapine is also proven to be safe, it has great potential to become a more desirable antipsychotic. [1] In summary, this study is the first to demonstrate the intrinsic effects of the most commonly used atypical antipsychotic on body weight, fat content, insulin sensitivity of the liver and peripheral tissues, and pancreatic β-cell function. The effects of olanzapine and rivaroxaban are significantly different. Olanzapine (OLZ) resulted in a significant increase in body weight, a significant increase in total trunk fat, reflecting significant expansion of visceral and subcutaneous adipose tissue, and severe hepatic insulin resistance. Rivaroxaban (RIS) had a smaller effect on fat, with no significant difference compared to the placebo group. More importantly, this study revealed that olanzapine significantly impaired the compensatory mechanism of β-cells in response to insulin resistance. Olanzapine completely blocked the compensatory response to insulin resistance induced by obesity and a high-fat diet, while β-cell function appeared to remain intact during rivaroxaban. The mechanisms of action of these antipsychotic drugs are not well understood, but these data suggest that these drugs may impair the neural regulation of β-cell compensation. The failure of β-cell compensation induced by atypical antipsychotic drugs provides a mechanistic explanation for the development of diabetes in vulnerable mental illness populations receiving such treatments. These results highlight the importance of studying drug effects after excluding common risk factors in patients with mental illness. Further research is needed to determine the potential mechanisms by which these drugs cause different metabolic sequelae and the processes that may lead to diabetes in this population. [5] |
| Molecular Formula |
C17H20N4S
|
|
|---|---|---|
| Molecular Weight |
312.44
|
|
| Exact Mass |
312.14
|
|
| Elemental Analysis |
C, 65.35; H, 6.45; N, 17.93; S, 10.26
|
|
| CAS # |
132539-06-1
|
|
| Related CAS # |
Olanzapine-d3; 786686-79-1; 132539-06-1; 783334-36-1 (HCl)
|
|
| PubChem CID |
135398745
|
|
| Appearance |
Yellow crystalline solid
Crystals from acetonitrile |
|
| Density |
1.3±0.1 g/cm3
|
|
| Boiling Point |
476.0±55.0 °C at 760 mmHg
|
|
| Melting Point |
195°C
|
|
| Flash Point |
241.7±31.5 °C
|
|
| Vapour Pressure |
0.0±1.2 mmHg at 25°C
|
|
| Index of Refraction |
1.709
|
|
| LogP |
2.18
|
|
| Hydrogen Bond Donor Count |
1
|
|
| Hydrogen Bond Acceptor Count |
4
|
|
| Rotatable Bond Count |
1
|
|
| Heavy Atom Count |
22
|
|
| Complexity |
432
|
|
| Defined Atom Stereocenter Count |
0
|
|
| SMILES |
S1C(C([H])([H])[H])=C([H])C2=C1N([H])C1=C([H])C([H])=C([H])C([H])=C1N=C2N1C([H])([H])C([H])([H])N(C([H])([H])[H])C([H])([H])C1([H])[H]
|
|
| InChi Key |
KVWDHTXUZHCGIO-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C17H20N4S/c1-12-11-13-16(21-9-7-20(2)8-10-21)18-14-5-3-4-6-15(14)19-17(13)22-12/h3-6,11,19H,7-10H2,1-2H3
|
|
| Chemical Name |
2-methyl-4-(4-methylpiperazin-1-yl)-10H-thieno[2,3-b][1,5]benzodiazepine
|
|
| Synonyms |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: This product requires protection from light (avoid light exposure) during transportation and storage. |
|
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
|
|||
|---|---|---|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2 mg/mL (6.40 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2 mg/mL (6.40 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. View More
Solubility in Formulation 3: ≥ 2 mg/mL (6.40 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2006 mL | 16.0031 mL | 32.0061 mL | |
| 5 mM | 0.6401 mL | 3.2006 mL | 6.4012 mL | |
| 10 mM | 0.3201 mL | 1.6003 mL | 3.2006 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
The Impact of Preoperative Olanzapine on Quality of Recovery After Discharge from Ambulatory Surgery
CTID: NCT05676294
Phase: Phase 2 Status: Recruiting
Date: 2024-10-08
|
|
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved





























 COA
COA