| Size | Price | Stock | Qty |
|---|---|---|---|
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg | |||
| Other Sizes |
































Purity: ≥98%
Norclozapine (ACP-104; NDMC; ACP104; N-Desmethylclozapine) is a major active metabolite of the atypical antipsychotic drug clozapine. It functions as a mild partial agonist at the D2 and D3 receptors, just like aripiprazole and bifeprunox, in contrast to clozapine, which lacks this intrinsic activity. It is a dopamine/muscarinic/5-HT2A inverse agonist that may be used to treat schizophrenia. N-desmethylclozapine functions as both an agonist and an inhibitor of the dengue virus receptor.
| Targets |
δ Opioid Receptor/DOR; mAChR1
|
|---|---|
| ln Vitro |
N-desmethylclozapine, a brain penetrant metabolite, was found to bind to M1 muscarinic receptors preferentially, with an IC50 of 55 nM. Compared to clozapine, it was a more potent partial agonist at this receptor, with an EC50 of 115 nM and 50% of acetylcholine response[1].
N-desmethylclozapine has agonistic characteristics at the 5-HT1A receptor in the cerebral cortex and hippocampus, as well as mild agonistic effects on the M1 mAChR. Additionally, this substance exhibits agonistic behavior at the δ-opioid receptor located in the striatum and cerebral cortex[2]. N-desmethylclozapine (3 μM) significantly reduces excitatory neurons' outward current, but not that of inhibitory neurons. When it comes to excitatory neurons, N-desmethylclozapine works better on its own than it does when combined with clozapine or taken alone. One microgram of atropine and 0.1 microgram of pirenzepine both considerably reduce the effects of N-desmethylclozapine on excitatory neurons. In excitatory cells, K125 channels were inhibited by N-desmethylclozapine but not by clozapine via M1 receptors[3]. N-desmethylclozapine causes TxB2 levels to drop both in response to TSST-1 stimulation and in unstimulated conditions. The production of TxA2 or TxB2 may be modulated by clozapine, N-desmethylclozapine, and CPZ, potentially affecting neurotransmitter systems[5]. N-desmethylclozapine, fluoxetine hydrochloride, and salmeterol xinafoate have IC50 values of 1 μM, 0.38 μM, and 0.67 μM in Huh-7 cells infected with DENV-2, respectively. When cells treated with all three inhibitors are compared to those treated with DMSO, the levels of NS3 are lower, indicating that the inhibitors function at a stage before the translation of viral proteins. Negative-strand RNA levels are reduced by >75% in cells treated with N-desmethylclozapine[6]. The atypical antipsychotic clozapine is widely used for treatment-resistant schizophrenic patients. Clozapine and its major active metabolite, N-desmethylclozapine (NDMC), have complex pharmacological properties, and interact with various neurotransmitter receptors. There are several biochemical studies reporting that NDMC exhibits a partial agonist profile at the human recombinant M1 muscarinic receptors. However, direct electrophysiological evidence showing the ability of NDMC to activate native M1 receptors in intact neurons is poor. Using rat hippocampal neurons, we previously demonstrated that activation of muscarinic receptors by a muscarinic agonist, oxotremorine M (oxo-M), induces a decrease in outward K(+)current at -40mV. In the present study, using this muscarinic current response we assessed agonist and antagonist activities of clozapine and NDMC at native muscarinic receptors in intact hippocampal excitatory and inhibitory neurons. Suppression of the oxo-M-induced current response by the M1 antagonist pirenzepine was evident only in excitatory neurons, while the M3 antagonist darifenacin was effective in both types of neurons. Muscarinic agonist activity of NDMC was higher than that of clozapine, higher in excitatory neurons than in inhibitory neurons, sensitive to pirenzepine, and partially masked when co-applied with clozapine. Muscarinic antagonist activity of clozapine as well as NDMC was not different between excitatory and inhibitory neurons, but clozapine was more effective than NDMC. These results demonstrate that NDMC has the ability to activate native M1 receptors expressed in hippocampal excitatory neurons, but its agonist activity might be limited in clozapine-treated patients because of the presence of excessive clozapine with muscarinic antagonist activity. [3] Thromboxane A2 (TxA2) and the activation of its receptor have been shown to modulate vasoconstriction and platelet aggregation as well as dopaminergic and serotonergic signalling. Dopaminergic and serotonergic systems play a crucial role in the pathophysiology of schizophrenia and these systems are the main targets of antipsychotics (APs). As the first antipsychotic (AP) chlorpromazine (CPZ) has already been shown to reduce TxA2, we hypothesized that the AP clozapine and its metabolite N-desmethylclozapine (NDMC) might also influence TxA2 production. We measured levels of thromboxane B2 (TxB2), the metabolite of the very unstable molecule TxA2, in unstimulated and stimulated blood samples of 10 healthy female subjects in a whole blood assay using toxic shock syndrome toxin-1 (TSST-1) and monoclonal antibody against surface antigen CD3 combined with protein CD40 (OKT3/CD40) as stimulants. Blood was supplemented with the APs CPZ, clozapine or NDMC in one of four different concentrations. Additionally, thromboxane levels were measured in blood without the addition of APs under different stimulation conditions. Under TSST-1 as well as OKT3/CD40 stimulation, mean TxB2 concentrations were significantly (p < 0.05) decreased by clozapine over all applied concentrations. NDMC led to a decrease in TxB2 levels under unstimulated conditions as well as under TSST-1 stimulation. CPZ reduced TxB2 production at low concentrations under unstimulated and TSST-1- stimulated conditions. Clozapine, NDMC and CPZ possibly act on neurotransmitter systems via modulation of TxA2 or TxB2 production. Additionally, known side effects of APs such as orthostatic hypotension may be a result of changes in the concentrations of TxA2 or TxB2.[5] Around 10,000 people die each year due to severe dengue disease, and two-thirds of the world population lives in a region where dengue disease is endemic. There has been remarkable progress in dengue virus vaccine development; however, there are no licensed antivirals for dengue disease, and none appear to be in clinical trials. We took the approach of repositioning approved drugs for anti-dengue virus activity by screening a library of pharmacologically active compounds. We identified N-desmethylclozapine, fluoxetine hydrochloride, and salmeterol xinafoate as dengue virus inhibitors based on reductions in the numbers of infected cells and viral titers. Dengue virus RNA levels were diminished in inhibitor-treated cells, and this effect was specific to dengue virus, as other flaviviruses, such as Japanese encephalitis virus and West Nile virus, or other RNA viruses, such as respiratory syncytial virus and rotavirus, were not affected by these inhibitors. All three inhibitors specifically inhibited dengue virus replication with 50% inhibitory concentrations (IC50s) in the high-nanomolar range. Estimation of negative-strand RNA intermediates and time-of-addition experiments indicated that inhibition was occurring at a postentry stage, most probably at the initiation of viral RNA replication. Finally, we show that inhibition is most likely due to the modulation of the endolysosomal pathway and induction of autophagy [6]. |
| ln Vivo |
N-desmethylclozapine underlies presynaptic modulation of glutamate release and GABA in humans and rats at M2 and M4 mAChRs, respectively. N-desmethylclozapine, for example, may be an M2 mAChR antagonist in rats, but it is not active at this receptor in the human neocortex. On the other hand, N-desmethylclozapine does not have an agonistic effect at M4 mAChR in the rat neocortex[4].
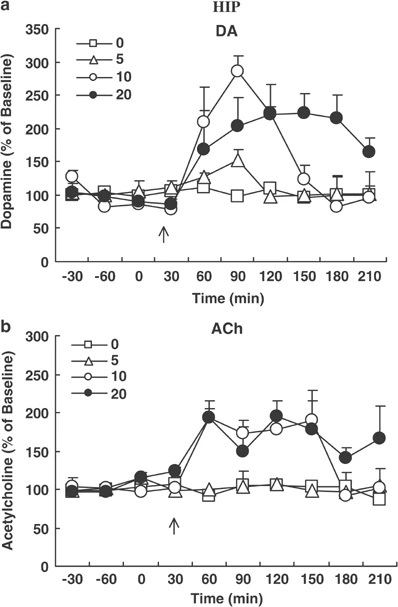
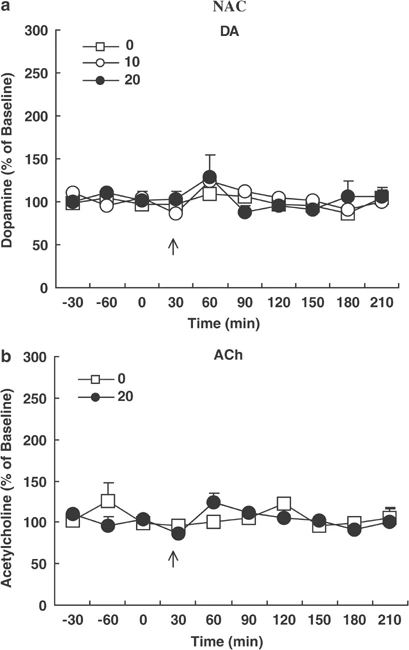
The active moiety of clozapine, the prototypical antipsychotic drug, consists of clozapine and its major metabolite, N-desmethylclozapine (NDMC). Previous studies have suggested that NDMC may be more important than the patent compound itself for the improvement in cognition in patients with schizophrenia treated with clozapine. While the pharmacology of clozapine and NDMC are similar in most respects, NDMC has been shown to be an M1 muscarinic receptor partial agonist whereas clozapine is an M1 antagonist in vitro and in vivo. We hypothesized that NDMC may improve cognition by increasing dopamine (DA) and acetylcholine (ACh) release in medial prefrontal cortex (mPFC) via direct stimulation of M1 receptors, whereas both NDMC and clozapine itself would do so by other mechanisms as well, and that clozapine would inhibit the M1 agonist effect of NDMC. In the present study, using microdialysis in awake, freely moving rats, we found that NDMC at doses of 10 and 20, but not 5 mg/kg, significantly increased DA and ACh release in the mPFC and HIP, but not in the nucleus accumbens (NAC). The M1-preferring antagonist, telenzepine (3 mg/kg), completely blocked NDMC (10 mg/kg)-induced increases in cortical DA and ACh release. Clozapine (1.25 mg/kg), which by itself had no effect on DA or ACh release in the cortex, blocked NDMC (10 mg/kg)-induced ACh, but not DA, release in the mPFC. The 5-HT1A receptor antagonist, WAY100635 (0.2 mg/kg) blocked NDMC (20 mg/kg)-induced cortical DA but not ACh release. These findings suggest that: (1) NDMC is an M1 agonist while clozapine is an M1 antagonist in vivo; (2) M1 agonism of NDMC can contribute to the release of cortical ACh and DA release; (3) NDMC, because of its M1 agonism, may more effectively treat the cognitive impairments observed in schizophrenia than clozapine itself; and (4) M1 receptor agonism may be a valuable target for the development of drugs that can improve cognitive deficit in schizophrenia, and perhaps other neuropsychiatric disorders as well. [1] Xanomeline had agonistic activity at the M1 muscarinic acetylcholine receptor (mAChR) in all brain regions, as well as at the 5-HT1A receptor in the cerebral cortex and hippocampus. On the other hand, N-desmethylclozapine exhibited slight agonistic effects on the M1 mAChR, and agonistic properties at the 5-HT1A receptor in the cerebral cortex and hippocampus. This compound also behaved as an agonist at the δ-opioid receptor in the cerebral cortex and striatum. In addition, the stimulatory effects of N-desmethylclozapine on [(35)S]GTPγS binding to Gαi/o were partially mediated through mAChRs (most likely M4 mAChR subtype), at least in striatum. Conclusions: The agonistic effects on the mAChRs (particularly M1 subtype, and also probably M4 subtype), the 5-HT1A receptor and the δ-opioid receptor expressed in native brain tissues, some of which are common to both compounds and others specific to either, likely shape the unique beneficial effectiveness of both compounds in the treatment for schizophrenic patients. These characteristics provide us with a clue to develop newer antipsychotics, beyond the framework of dopamine D2 receptor antagonism, that are effective not only on positive symptoms but also on negative symptoms and/or cognitive/affective impairment.[2] Cholinergic transmission plays a pivotal role in learning, memory and cognition, and disturbances of cholinergic transmission have been implicated in neurological disorders including Alzheimer's disease, epilepsy and schizophrenia. Pharmacological alleviation of these diseases by drugs including N-desmethylclozapine (NDMC), promising in animal models, often fails in patients. We therefore compared the effects of NDMC on glutamatergic and GABAergic transmission in slices from rat and human neocortex. We used carbachol (CCh; an established agonist at metabotropic muscarinic acetylcholine (ACh) receptors (mAChRs)) as a reference. Standard electrophysiological methods including intracellular and field potential recordings were used. In the rat neocortex, NDMC prevented the CCh-induced decrease of GABAA and GABAB receptor-mediated responses but not the CCh-induced increase of the paired-pulse depression. NDMC reduced neither the amplitude of the excitatory postsynaptic potentials (EPSP) nor antagonized the CCh-induced depression of EPSP. In the human neocortex, however, NDMC failed to prevent CCh-induced decrease of the GABAB responses and directly reduced the amplitude of EPSP. These data suggest distinct effects of NDMC in rat and human at M2 and M4 mAChRs underlying presynaptic modulation of GABA and glutamate release, respectively. In particular, NDMC might be a M2 mAChR antagonist in the rat but has no activity at this receptor in human neocortex. However, NDMC has an agonistic effect at M4 mAChR in the human but no such effect in the rat neocortex. The present study confirms that pharmacology at mAChRs can differ between species and emphasizes the need of studies in human tissue [4]. |
| Cell Assay |
Inhibitor treatments. [6]
Dengue virus infection and inhibitor treatment experiments were performed with Huh-7 and A549 cells, and viral titers in the culture supernatants were determined by plaque assays on BHK-21 cells. IC50 (50% inhibitory concentration) experiments were performed by using 2-fold dilutions of the inhibitor starting from an 8 μM concentration. A total of 30,000 cells were plated in a 48-well plate, infected with DENV-2 at a multiplicity of infection (MOI) of 3, and incubated with 2% Dulbecco's modified Eagle's medium (DMEM) containing inhibitors. Supernatants were collected at 24 h postinfection (p.i.), and viral titers were estimated by plaque assays on BHK-21 cells. For Western blotting, cell lysates were prepared as described previously (10), under the inhibitor treatment conditions described below, and the membranes were probed with antibodies recognizing DENV nonstructural protein 3 (NS3) (kind gift from Raj Bhatnagar), tubulin monoclonal, calnexin, and 78-kDa glucose-regulated protein (GRP78) antibodies. For LC3 detection, an antibody that has a high specificity for the LC3-II form was used (LC3b [D11]; Cell Signaling Technology). Signals were detected by chemiluminescence. (See the supplemental material for a description of the flow cytometry experiment.) |
| Animal Protocol |
Drugs [1]
NDMC/N-desmethylclozapine and clozapine was dissolved in a small amount of 0.1 M tartaric acid and the pH was adjusted to 6–7 with 0.1 N NaOH. WAY100635 (Wyeth Laboratories, Philadelphia, PA) and telenzepine (Research Chemical Inc.) were dissolved in deionized water. Vehicle or drugs in a volume of 1.0 ml/kg were administered subcutaneously to randomly assigned rats. At 3–5 days after cannulation, a dialysis probe was implanted into the mPFC and NAC under slight anesthesia with isoflurane. Rats were then housed individually overnight in a dialysis cage. After the overnight perfusion at 0.4 μl/min of the probe, the flow was increased to 1.5 μl/min. After 1 h, the dialysate samples were collected every 30 min. The perfusion medium was Dulbecco's phosphate-buffered saline solution, including Ca2+ (138 mM NaCl, 8.1 mM Na2HPO4, 2.7 mM KCl, 1.5 mM KH2PO4, 0.5 mM MgCl, 1.2 mM CaCl2, pH 7.4). No AChesterase inhibitor in the dialysate is required with this procedure (Ichikawa et al, 2002b). After stable baseline values in the dialysates were obtained, each rat received two injections, vehicle/NDMC (N-desmethylclozapine), WAY100635/NDMC, telenzepine/NDMC, or clozapine/NDMC. The locations of the dialysis probes were verified at the end of each experiment by brain dissection. Background: 3(3-Hexyloxy-1,2,5-thiadiazol-4-yl)-1,2,5,6-tetrahydro-1-methylpyridine (xanomeline) and N-desmethylclozapine are of special interest as promising antipsychotics with better efficacy, especially for negative symptoms and/or cognitive/affective impairment. Methods: The guanosine-5'-O-(3-[(35)S]thio)triphosphate ([(35)S]GTPγS) binding experiments were performed using (1) conventional filtration technique, (2) antibody-capture scintillation proximity assay, and (3) immunoprecipitation method, in brain membranes prepared from rat cerebral cortex, hippocampus, and striatum.[2] |
| ADME/Pharmacokinetics |
Metabolism / Metabolites
Known metabolites of N-desmethylclozapine include desmethylclozapine N-glucuronide. N-desmethylclozapine is a known metabolite of clozapine. |
| References |
|
| Additional Infomation |
N-Desmethylclozapine is a dibenzodiazepine compound containing chlorine and piperazine groups in its molecular structure and is the major metabolite of clozapine. Clozapine is a potent and selective 5-HT2C serotonin receptor antagonist. It is both a metabolite of clozapine and a delta-opioid receptor agonist and a serotonergic antagonist. It belongs to the dibenzodiazepine class, piperazine class, and organochlorine class of compounds. ACP-104, or N-desmethylclozapine, is the major metabolite of clozapine and is currently being developed by ACADIA as a novel standalone therapy for schizophrenia. It combines the therapeutic characteristics of atypical antipsychotics with the potential benefits of enhancing cognitive function, thus addressing a major challenge in the treatment of schizophrenia today.
Drug Indications Studied for the treatment of schizophrenia and schizoaffective disorder. Mechanism of Action ACP-104 combines an M1 muscarinic receptor agonist, a 5-HT2A receptor inverse agonist, and partial agonists of D2 and D3 dopamine receptors in a single compound. ACP-104 uniquely stimulates brain cells called M1 muscarinic receptors, which play a crucial role in cognition. ACP-104 is a partial agonist that weakly activates dopamine D2 and D3 receptors. These partial agonist properties of ACP-104 may result in fewer motor side effects compared to most other antipsychotics. Pharmacodynamics ACP-104 is a small molecule drug candidate we are developing for a novel therapy to treat schizophrenia. It is known that after taking clozapine, a large amount of ACP-104, namely N-desmethylclozapine, is generated in the body. That is, clozapine is metabolized into ACP-104. We discovered that ACP-104 possesses a unique ability to stimulate the m1 muscarinic receptor, a key muscarinic receptor. The m1 muscarinic receptor is known to play a crucial role in cognition. Since clozapine itself blocks the m1 muscarinic receptor, patients need to metabolize a significant amount of clozapine into ACP-104 to stimulate this receptor, thereby overcoming clozapine's blocking effect. Using ACP-104 avoids the variability of metabolic processes and the competitive effects of clozapine. Like clozapine, ACP-104 is a dopamine antagonist and a 5-HT2A inverse agonist. We believe that ACP-104 represents a novel approach to treating schizophrenia, combining the therapeutic characteristics of atypical antipsychotics with the added advantage of beneficial cognitive effects. The main findings of this study are: (1) NDMC, the main active metabolite of clozapine, significantly increased the release of dopamine (DA) and acetylcholine (ACh) in the medial prefrontal cortex (mPFC) and hippocampus (HIP), but had no effect on the nucleus accumbens (NAC); (2) tilenzapine, a preferential M1 receptor antagonist, completely blocked the release of DA and ACh in the mPFC induced by NDMC; (3) Clozapine (1.25 mg/kg) completely blocked the release of ACh induced by NDMC (10 mg/kg), which is consistent with previous reports that NDMC is a potent M1 receptor agonist, while clozapine has the characteristics of an M1 receptor antagonist in vivo; (4) Clozapine pretreatment did not block the release of cortical DA induced by NDMC, indicating that M1 receptor agonism is not the reason for this effect of NDMC; (5) 5-HT1A The receptor antagonist WAY100635 partially blocked the increase in DA (but not ACh) release from the mPFC induced by NDMC, indicating that cortical DA release is partially dependent on stimulation of the 5-HT1A receptor. The effect of clozapine on DA or ACh release is likely a result of the combined effect of clozapine and NDMC, i.e., a mixed agonist/antagonist effect. Therefore, high levels of NDMC, especially a high NDMC/clozapine ratio, enhance the stimulation of M1 muscarinic receptors, consistent with predictions from mass action theory and mixed agonist/antagonist experiments (Brauner-Osborne et al., 1996). It has been reported that clozapine concentrations in the brain of rats during long-term clozapine administration were three times higher than NDMC concentrations (Weigmann et al., 1999). Currently, there is no information on the relative levels of NDMC and clozapine in humans. High concentrations of NDMC have been detected in plasma samples from some patients treated with clozapine (Hasegawa et al., 1993). High levels of NDMC, especially a high NDMC/clozapine ratio, further enhance the stimulatory effect on M1 muscarinic receptors. Current data on clozapine blocking NDMC-induced ACh release are consistent with our laboratory's clinical data, which suggest that the NDMC/clozapine ratio is a better predictor of clozapine's clinical efficacy than clozapine concentration alone (Frazier et al., 2003; Mauri et al., 2003; Weiner et al., 2004). In summary, NDMC preferentially increases DA and ACh release in mPFC and HIP, but not in NAC, similar to the effects of clozapine and other atypical antipsychotics. The blocking of NDMC-induced ACh release by tirizine and clozapine indicates that M1 receptor stimulation contributes to NDMC's increased cortical DA and ACh release, confirming that NDMC has a significant M1 receptor agonist effect, while its parent compound clozapine is an antagonist. [1] In this study, we assessed the agonist and antagonist activities of clozapine and its major active metabolite NDMC on native muscarinic receptors in intact hippocampal excitatory and inhibitory neurons in rats by monitoring muscarinic current responses. Our data clearly show that clozapine primarily acts as an antagonist, while NDMC acts as a mixed agonist-antagonist. The agonist activity of NDMC (which could be explained by its action on M1 rather than M3 subtype receptors) was largely masked by simultaneous administration of the same concentration of clozapine. These results suggest that the clozapine/NDMC concentration ratio in clozapine-treated patients may determine the extent to which NDMC activates M1 receptors. [3] In conclusion, the current data raise some questions about extrapolating findings from rat neurons to human neurons, especially when using drugs other than standard compounds, as the pharmacological properties of NDMC differ significantly between the two species (Thomas et al., 2010). Therefore, despite the limitations of human tissues themselves, this study underscores the need to conduct research in human tissues. [4] |
| Molecular Formula |
C17H17CLN4
|
|---|---|
| Molecular Weight |
312.797
|
| Exact Mass |
312.114
|
| Elemental Analysis |
C, 65.28; H, 5.48; Cl, 11.33; N, 17.91
|
| CAS # |
6104-71-8
|
| Related CAS # |
N-Desmethylclozapine-d8; 1189888-77-4; N-Desmethylclozapine-d8 hydrochloride; 2705402-91-9
|
| PubChem CID |
135409468
|
| Appearance |
Light yellow to green yellow solid powder
|
| Density |
1.38g/cm3
|
| Boiling Point |
490.1ºC at 760 mmHg
|
| Melting Point |
120-125°C
|
| Flash Point |
250.2ºC
|
| Index of Refraction |
1.709
|
| LogP |
3.22
|
| Hydrogen Bond Donor Count |
2
|
| Hydrogen Bond Acceptor Count |
3
|
| Rotatable Bond Count |
1
|
| Heavy Atom Count |
22
|
| Complexity |
421
|
| Defined Atom Stereocenter Count |
0
|
| SMILES |
ClC1=CC=C2C(N=C(N3CCNCC3)C4=CC=CC=C4N2)=C1
|
| InChi Key |
JNNOSTQEZICQQP-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C17H17ClN4/c18-12-5-6-15-16(11-12)21-17(22-9-7-19-8-10-22)13-3-1-2-4-14(13)20-15/h1-6,11,19-20H,7-10H2
|
| Chemical Name |
3-chloro-6-piperazin-1-yl-11H-benzo[b][1,4]benzodiazepine
|
| Synonyms |
AZD-5991; AZD-5991 S-enantiomer; N-Desmethylclozapine; Norclozapine; 6104-71-8; Desmethylclozapine; N-desmethyl clozapine; N-desmethyl-clozapine; N-DEMETHYLCLOZAPINE; 8-Chloro-11-(1-piperazinyl)-5H-dibenzo(b,e)(1,4)diazepine; AZD 5991 S-enantiomer.
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO: ≥ 50 mg/mL (~159.9 mM)
|
|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (7.99 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (7.99 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1969 mL | 15.9847 mL | 31.9693 mL | |
| 5 mM | 0.6394 mL | 3.1969 mL | 6.3939 mL | |
| 10 mM | 0.3197 mL | 1.5985 mL | 3.1969 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00628420 | Completed | Drug: ACP-104 Drug: Placebo |
Schizophrenia | University of Texas Southwestern Medical Center |
January 2005 | Phase 1 |
| NCT00490516 | Completed | Drug: ACP-104 Drug: Placebo |
Schizophrenia | ACADIA Pharmaceuticals Inc. | June 2007 | Phase 2 |
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved





























 COA
COA