| Size | Price | Stock | Qty |
|---|---|---|---|
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g | |||
| Other Sizes |
































Purity: ≥98%
Lovastatin (Mevinolin; Mevacor; MK-803; Altoprev; Monacolin K; MK803), belonging to the statin class of hypolipidemic agent/lipid-lowering drugs, is a potent and cell-permeable inhibitor of HMG-CoA reductase with potential anti-hyperlipidemic effects. It inhibits HMG-CoA reductase with an IC50 of 3.4 nM in a cell-free assay. Lovastatin has been approved for use in reducing/lowering cholesterol.
| Targets |
Selective inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase (rate-limiting enzyme in cholesterol biosynthesis) with the following inhibitory parameters:
- Ki = 10 nM (purified rat liver HMG-CoA reductase), showing high affinity for the enzyme [1] - IC50 = 25 nM (human recombinant HMG-CoA reductase); inhibits enzyme activity by >90% at 100 nM [3] |
|---|---|
| ln Vitro |
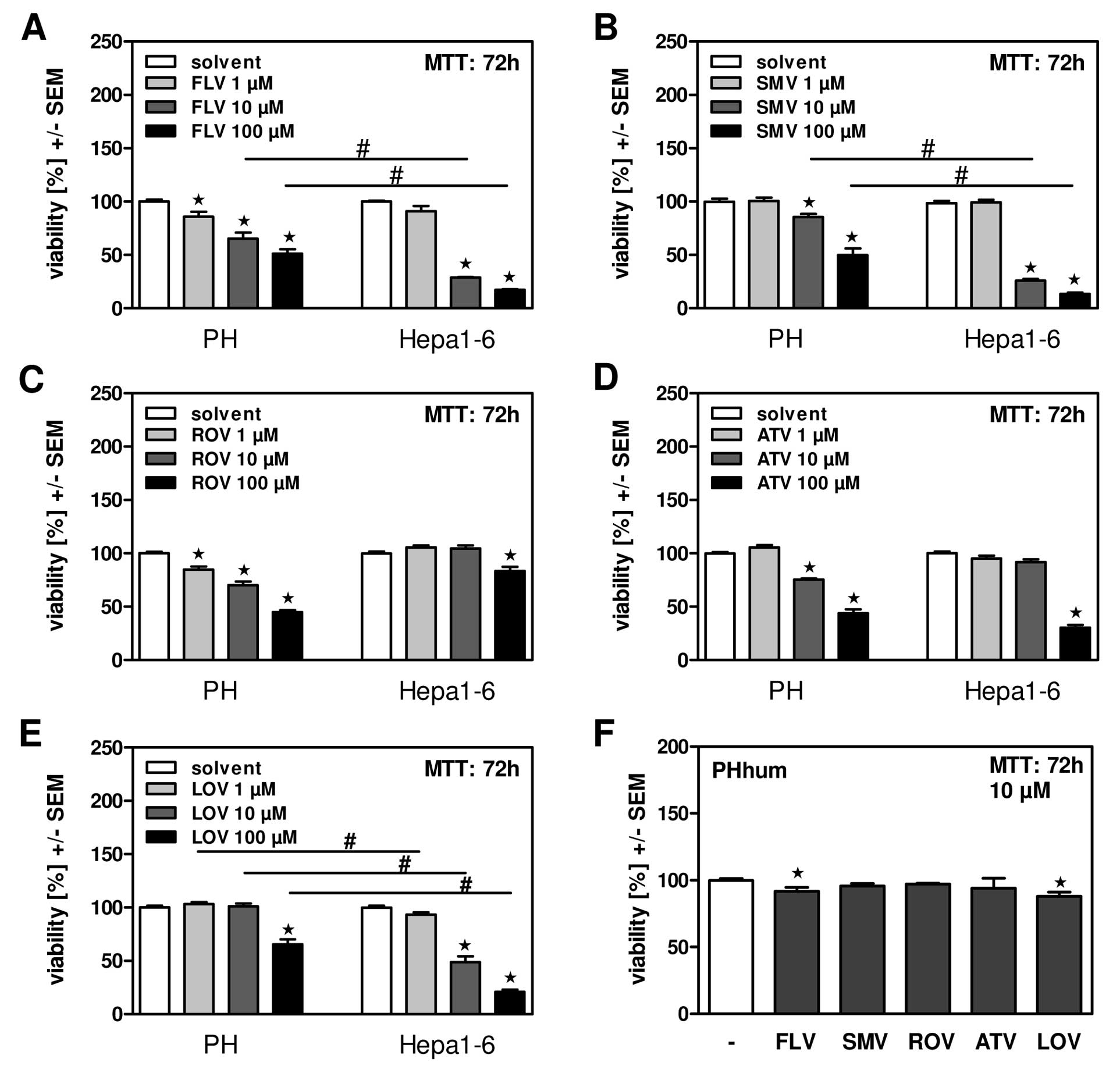
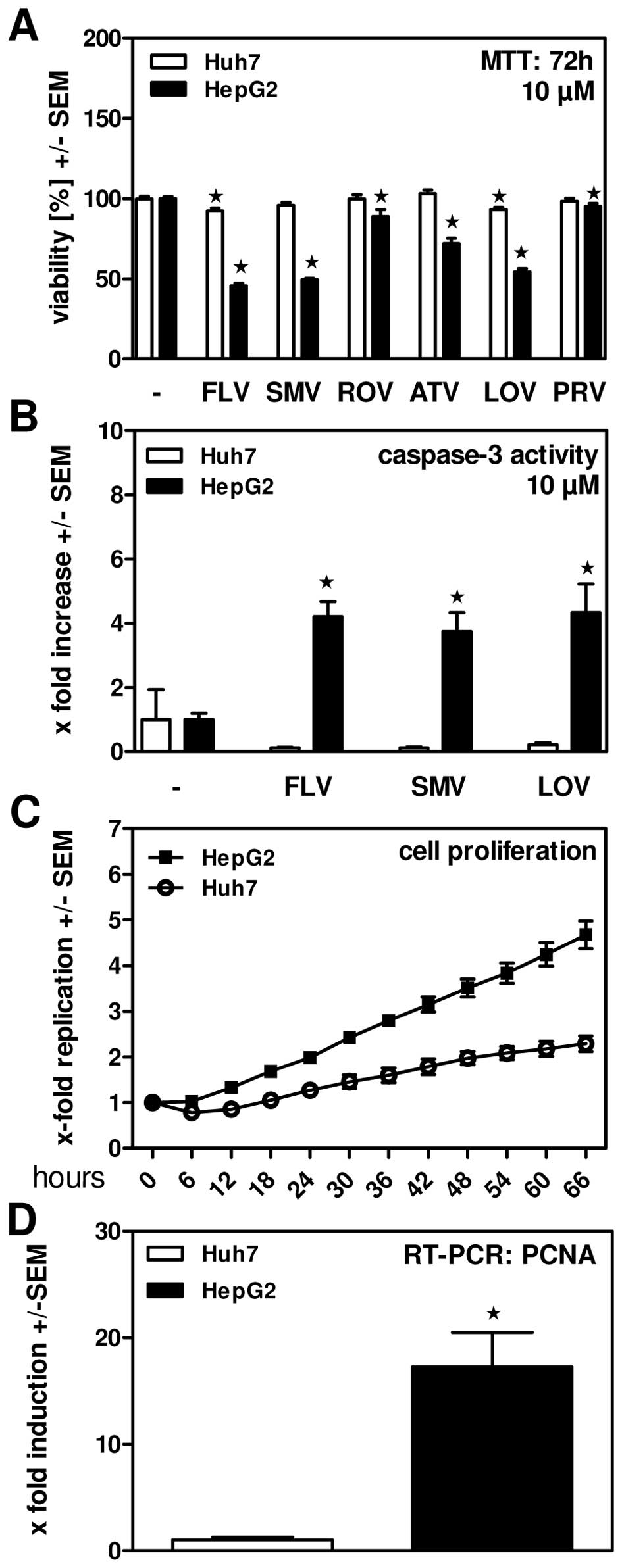
HepG2 cell viability is effectively reduced by lovastatin (10 μM; 72 hours)[2]. In HepG2 cells, lovastatin (10 μM; 48 hours) causes apoptosis [2].
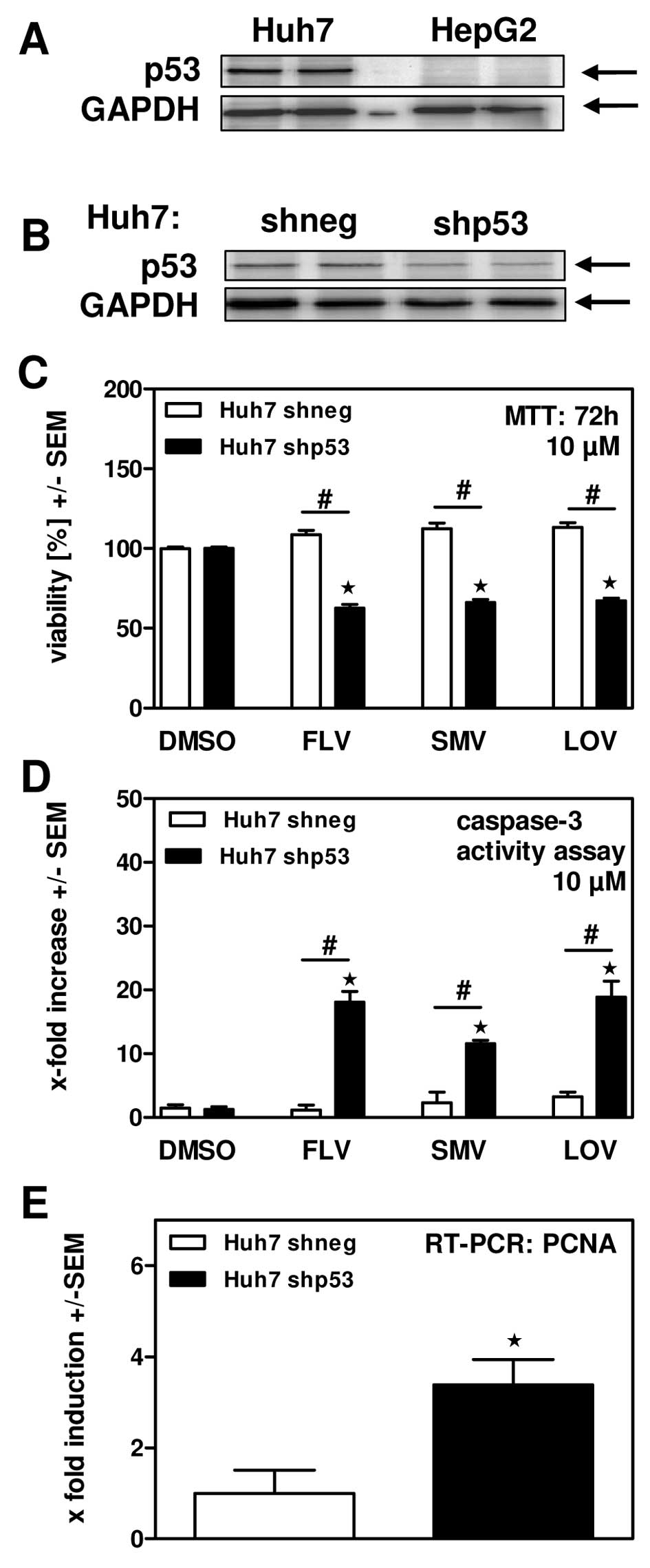
Inhibition of HMG-CoA reductase and cholesterol synthesis: - In purified rat liver HMG-CoA reductase assays, Lovastatin (1–100 nM) inhibited enzyme activity in a concentration-dependent manner: - 10 nM inhibited 50% of activity (consistent with Ki=10 nM); - 100 nM inhibited >90% of activity, blocking conversion of HMG-CoA to mevalonate [1] - In primary human hepatocytes, Lovastatin (0.1–10 μM, 24-hour treatment) reduced de novo cholesterol synthesis: - 1 μM decreased [14C]-acetate incorporation into cholesterol by 40%; - 10 μM decreased cholesterol synthesis by 75%, with no significant cytotoxicity (>90% viability via MTT assay) [3] - Induction of apoptosis in hepatoma cells (p53-dependent): - In human hepatoma cells: - p53-wild-type cells (HepG2): Lovastatin (5–50 μM, 72-hour treatment) inhibited proliferation with IC50=15 μM (MTT assay); 25 μM increased Annexin V-positive apoptotic cells from 4% to 38% (flow cytometry) [2] - p53-mutant cells (Hep3B): Minimal effect—50 μM Lovastatin only reduced viability by 20% and induced apoptosis in 8% of cells, confirming p53 dependence [2] - Mechanistic effects (25 μM Lovastatin, HepG2 cells): - Upregulated p53 protein by 2.1-fold (Western blot); - Increased cleaved caspase-3 by 3.2-fold and Bax/Bcl-2 ratio by 2.5-fold; - Reduced mitochondrial membrane potential by 45% (JC-1 staining) [2] - Reduction of blood-brain barrier (BBB) permeability and leukocyte migration: - In human brain microvascular endothelial cells (HBMECs), Lovastatin (1–10 μM, 24-hour treatment): - 10 μM reduced BBB permeability (measured via fluorescein isothiocyanate-dextran flux) by 35% [5] - In human peripheral blood mononuclear cells (PBMCs), 10 μM Lovastatin inhibited migration toward CCL2 (a chemokine) by 50% (Transwell assay) [5] |
| ln Vivo |
The liver hydrolyzes the inactive lactone lovastatin to produce the active f3-hydroxy acid form. This primary metabolite inhibits the enzyme HMG-CoA reductase. Ki is one nanometer. Human plasma proteins bind lovastatin and its beta-hydroxy acid metabolite tightly. Both the placental and blood-brain barriers are crossed by lovastatin [3]. Lovastatin modestly raises HDL cholesterol while considerably lowering apolipoprotein B-containing lipoproteins, particularly LDL cholesterol, and plasma triglycerides to a lesser level [4].
Lipid-lowering efficacy in hypercholesterolemic animal models: 1. High-cholesterol diet (HCD)-fed rabbits (New Zealand white rabbits, 2–3 kg): - Rabbits were randomized into 3 groups (n=6/group): vehicle (0.5% CMC-Na), Lovastatin 1 mg/kg/day, 5 mg/kg/day [1] - Treatment: Daily oral gavage for 28 days (continued HCD). Fasting serum samples were collected weekly [1] - Results: - Serum low-density lipoprotein cholesterol (LDL-C): 5 mg/kg group reduced LDL-C by 45% vs. vehicle (vehicle LDL-C: 320 ± 40 mg/dL) on day 28; - Serum total cholesterol (TC): 5 mg/kg group reduced TC by 38% vs. vehicle (vehicle TC: 450 ± 50 mg/dL) [1] 2. HCD-fed rats (Sprague-Dawley rats, 250–300 g): - Oral Lovastatin 10 mg/kg/day for 21 days reduced serum LDL-C by 35% and TC by 30% vs. vehicle; serum high-density lipoprotein cholesterol (HDL-C) increased by 12% [3] - Efficacy in experimental autoimmune encephalomyelitis (EAE) mice (multiple sclerosis model): 1. Animals: C57BL/6 mice (8 weeks old) were immunized with myelin oligodendrocyte glycoprotein (MOG35-55) peptide to induce EAE [5] 2. Treatment: Lovastatin 20 mg/kg/day (dissolved in 0.5% CMC-Na) was administered via oral gavage from day 0 to day 21 post-immunization [5] 3. Results: - BBB permeability: Lovastatin reduced Evans blue extravasation into the brain by 40% vs. vehicle; - Leukocyte infiltration: Spinal cord leukocyte count decreased by 50% (flow cytometry); - Clinical EAE score: Reduced from 3.2 ± 0.5 (vehicle) to 1.5 ± 0.3 (treated group) [5] |
| Enzyme Assay |
Purified rat liver HMG-CoA reductase activity assay :
The reaction system (250 μL) contained 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 2 mM DTT, 50 μg purified rat liver HMG-CoA reductase, 15 μM [14C]-HMG-CoA (substrate), 200 μM NADPH (cofactor), and Lovastatin (0.1–100 nM). The mixture was incubated at 37°C for 90 minutes. The reaction was terminated by adding 100 μL of 6 M HCl, followed by heating at 100°C for 15 minutes to convert mevalonate (product) to mevalonolactone. Mevalonolactone was extracted with 500 μL ethyl acetate, and the organic phase was transferred to a scintillation vial. Radioactivity was measured via liquid scintillation counting. Inhibition rate was calculated by comparing with the vehicle group, and Ki was determined using Lineweaver-Burk plot analysis (varying [14C]-HMG-CoA concentrations: 5–30 μM) [1] |
| Cell Assay |
Cell Viability Assay[2]
Cell Types: HepG2 cells Tested Concentrations: 10 μM Incubation Duration: 72 hrs (hours) Experimental Results: Efficiently decreased viability of HepG2 cells. Hepatoma cell proliferation and apoptosis assay : 1. Cell culture: HepG2 (p53-wild-type) and Hep3B (p53-mutant) cells were seeded in 96-well plates (5×103 cells/well) or 6-well plates (2×105 cells/well) in DMEM medium (10% FBS) at 37°C, 5% CO2 for 24 hours [2] 2. Drug treatment: Lovastatin (5–50 μM, dissolved in 0.1% DMSO) was added, and cells were incubated for 48–72 hours. Vehicle group received 0.1% DMSO [2] 3. Proliferation assay (96-well plates): MTT solution (5 mg/mL) was added for 4 hours; formazan crystals were dissolved with DMSO, and absorbance at 570 nm was measured. IC50 was calculated via non-linear regression [2] 4. Apoptosis assay (6-well plates): Cells were stained with Annexin V-FITC/PI for 15 minutes at room temperature, and apoptotic cells were quantified via flow cytometry. Mitochondrial membrane potential was assessed with JC-1 dye (red/green fluorescence ratio) [2] 5. Western blot: Cells were lysed with RIPA buffer (含protease inhibitors); 30 μg protein was separated by 10% SDS-PAGE, transferred to PVDF membranes, and probed with antibodies against p53, cleaved caspase-3, Bax, Bcl-2, and β-actin (loading control) [2] - BBB permeability and leukocyte migration assay : 1. HBMEC culture: HBMECs were seeded on Transwell inserts (polycarbonate membrane, 0.4 μm pore size) and cultured until confluent (7 days) [5] 2. Permeability assay: Lovastatin (1–10 μM) was added to the upper chamber for 24 hours. Fluorescein isothiocyanate-dextran (4 kDa) was added to the upper chamber, and fluorescence intensity in the lower chamber was measured after 2 hours to calculate permeability coefficient (Papp) [5] 3. PBMC migration assay: PBMCs were isolated from healthy donors and labeled with Calcein-AM. Lovastatin (1–10 μM) was pre-incubated with PBMCs for 1 hour; cells were added to the upper Transwell chamber, and CCL2 (100 ng/mL) was added to the lower chamber. After 4 hours, migrated cells in the lower chamber were counted via fluorescence microscopy [5] |
| Animal Protocol |
10 mg/kg
Mice HCD-fed hypercholesterolemic rabbit study : 1. Animals: New Zealand white rabbits (2–3 kg, male) were housed under 12-hour light/dark cycle (22±2°C) and fed a HCD (0.5% cholesterol, 10% lard) for 2 weeks to induce hypercholesterolemia [1] 2. Grouping: Rabbits were randomized into 3 groups (n=6/group): - Vehicle group: 0.5% carboxymethyl cellulose sodium (CMC-Na) solution; - Lovastatin 1 mg/kg/day group; - Lovastatin 5 mg/kg/day group [1] 3. Drug preparation: Lovastatin was dissolved in DMSO (5% v/v) and diluted with 0.5% CMC-Na to final concentration (DMSO ≤5%), sonicated for 5 minutes to ensure homogeneity [1] 4. Administration: Daily oral gavage at a volume of 5 mL/kg for 28 days (rabbits continued on HCD). Rabbits were fasted for 8 hours before weekly serum collection [1] 5. Sample detection: Serum LDL-C, TC, and HDL-C were quantified via enzymatic kits [1] - EAE mouse model study : 1. EAE induction: C57BL/6 mice (8 weeks old, female) were immunized subcutaneously with 200 μg MOG35-55 peptide emulsified in complete Freund’s adjuvant (CFA) containing 500 μg heat-killed Mycobacterium tuberculosis. Pertussis toxin (200 ng) was administered intraperitoneally on day 0 and day 2 post-immunization [5] 2. Grouping: Mice were randomized into 2 groups (n=8/group): vehicle (0.5% CMC-Na), Lovastatin 20 mg/kg/day [5] 3. Drug administration: Daily oral gavage from day 0 to day 21 post-immunization (volume: 10 mL/kg) [5] 4. Sample collection and detection: - Clinical scoring: Daily assessment of EAE severity (0=normal, 1=tail limpness, 2=hind limb weakness, 3=hind limb paralysis, 4=tetraplegia); - BBB permeability: Evans blue (2% in saline) was injected intravenously on day 21; 2 hours later, brains were homogenized, and Evans blue was extracted with formamide to measure absorbance at 620 nm; - Leukocyte infiltration: Spinal cords were dissected, digested, and leukocytes were counted via flow cytometry [5] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion
Lovastatin's Cmax is 3.013 ng/mL, and Tmax is 3.36 hours. The plasma concentration of total radioactivity (lovastatin plus its ¹⁴C metabolite) peaks 2 hours after administration and rapidly declines, reaching approximately 10% of the peak concentration by 24 hours. In four animal models, the average estimated absorption of lovastatin, based on the intravenously administered reference dose, is approximately 30% of the oral dose. Animal studies have shown that lovastatin exhibits high hepatic selectivity after oral administration, with significantly higher concentrations in the liver than in non-target tissues. Lovastatin undergoes extensive first-pass metabolism in its primary site of action—the liver—followed by bile excretion as an equivalent. Due to the extensive extraction of lovastatin from the liver, the concentrations entering systemic circulation are low and highly variable. In a single-dose study of four patients with hypercholesterolemia, it was estimated that less than 5% of the drug entered systemic circulation as an inhibitor of activity after oral administration of lovastatin. Following administration of lovastatin tablets, the coefficient of variation (AUC) for the total systemic circulating inhibitory activity, based on inter-subject variability, was approximately 40%. Peak concentrations of lovastatin after administration of 10–40 mg have been reported to range from 1.04–4.03 ng/ml, with AUCs of 14–53 ng·h/ml. This indicates that lovastatin exhibits dose-dependent pharmacokinetic characteristics. When lovastatin is taken on an empty stomach, the average plasma concentrations of both the active inhibitor and the total inhibitor are approximately two-thirds of those when taken immediately after a meal. Genetic differences in the hepatic transporter OATP1B1 (organic anion transport polypeptide 1B1), encoded by the SCLCO1B1 gene (a member of the solute carrier organic anion transporter family 1B1), have been shown to affect the pharmacokinetics of lovastatin. Pharmacogenetic studies of the c.521T>C single nucleotide polymorphism (SNP) showed that the lovastatin Cmax and AUC were 340% and 286% higher, respectively, compared to those homozygous for 521TT. The 521CC genotype was also associated with a significantly increased risk of myopathy, likely due to increased systemic exposure. Other statins affected by this polymorphism include rosuvastatin, pitavastatin, atorvastatin, simvastatin, and pravastatin. Although specific dosage instructions are not included in existing lovastatin product information, individuals carrying the c.521CC OATP1B1 genotype should be monitored for the risk of adverse reactions due to increased drug exposure, such as muscle pain and rhabdomyolysis, especially at high doses. After oral administration of c.521C-labeled lovastatin, 10% of the dose is excreted in the urine and 83% in the feces. The latter represents the amount of absorbed drug excreted along with unabsorbed drug via bile. Lovastatin can cross the blood-brain barrier and the placenta. /Breast Milk/ It is unclear whether lovastatin is excreted into human breast milk. Following oral administration of 14C-labeled lovastatin in humans, 10% of the dose is excreted in urine and 83% in feces. The latter represents the amount of absorbed drug excreted via bile, along with any unabsorbed drug. The total plasma radioactivity (lovastatin plus 14C metabolites) peaks at 2 hours post-administration and rapidly declines to approximately 10% of the peak at 24 hours post-administration. In four test animals, the average absorption rate of lovastatin (estimated relative to the intravenous reference dose) was approximately 30% of the oral dose. Animal studies have shown that lovastatin is highly selective for the liver after oral administration, with significantly higher concentrations in the liver than in non-target tissues. Lovastatin undergoes extensive first-pass metabolism in its primary site of action—the liver—followed by bile excretion. Due to the extensive extraction of lovastatin in the liver, its systemic circulation concentrations are low and highly variable. In a single-dose study of four patients with hypercholesterolemia, it was estimated that less than 5% of the drug entered systemic circulation as an inhibitor of activity after oral administration of lovastatin. The coefficient of variation for the area under the curve (AUC) of total systemic inhibitory activity, based on inter-subject variability, after administration of lovastatin tablets was approximately 40%. Lovastatin and its β-hydroxy acid metabolites are highly bound to human plasma proteins (>95%). Animal studies have shown that lovastatin can cross the blood-brain barrier and the placental barrier. Peak plasma concentrations of both the inhibitor of activity and the total inhibitor of activity are reached within 2 to 4 hours after administration. Lovastatin is administered as a lactone prodrug; therefore, it needs to be converted to its active β-hydroxy form to exert its mechanism of action. The activation process of this drug appears to be independent of CYP isoenzyme activity, but rather regulated by serum paraoxygenase activity. Lovastatin is metabolized by the hepatic microsomal enzyme system (cytochrome P-450 isoenzyme 3A4). The main active metabolites in human plasma are the β-hydroxy acid of lovastatin and its 6'-hydroxy, 6'-hydroxymethyl, and 6'-methylene derivatives. OATP1B1 activity enhances hepatic uptake of lovastatin. The main active metabolites of lovastatin in human plasma are the β-hydroxy acid and its 6'-hydroxy, 6'-hydroxymethyl, and 6'-methylene derivatives. The main active metabolites of lovastatin in human plasma are the β-hydroxy acid, its 6'-hydroxy derivative, and two other metabolites. Known metabolites of lovastatin include 3-hydroxylovastatin and 6'β-hydroxylovastatin. Lovastatin is metabolized in the liver, and its main active metabolites are the β-hydroxy acid, its 6'-hydroxy derivative, and two other metabolites. Excretion route: Lovastatin undergoes extensive first-pass metabolism in the liver, its primary site of action, followed by bile excretion. 83% of the oral dose is excreted in bile, and 10% in urine. Half-life: 5.3 hours Biological half-life The half-life of lovastatin has been reported to be 13.37 hours. The elimination half-life of the hydroxy acid form of lovastatin has been reported to be 0.7–3 hours. Oral absorption (References [3], [4]): - - Healthy volunteers: A single oral dose of 40 mg lovastatin (tablets) showed low oral bioavailability (F), 5%–10% (due to extensive first-pass metabolism in the liver and poor solubility); time to reach maximum concentration (Tmax) was 2–4 hours; maximum plasma concentration (Cmax) = 10–20 ng/mL (active β-hydroxy acid form) [3][4] - Metabolism (References [3], [4]): - Hepatic metabolism: Lovastatin is a prodrug—converted to its active β-hydroxy acid form by hepatic esterases; further metabolized by cytochrome P450 (CYP) 3A4 to an inactive metabolite (e.g., 6'-hydroxylovastatin) [3][4] - Elimination: - Elimination half-life (t1/2) = 1.5–2 hours (active form, healthy volunteers); - Excretion: 83% of the dose is excreted in feces (60% as metabolites and 23% as the original drug), and 10% is excreted in urine [3] - Distribution: - Volume of distribution (Vd) = 134 L (healthy volunteers, 40 mg orally); - High concentration in the liver (target organ) - liver/plasma concentration ratio = 10:1 [3] |
| Toxicity/Toxicokinetics |
Toxicity Summary
Identification and Uses: Lovastatin is a white, non-hygroscopic crystalline powder. It is used as a cholesterol-lowering drug and an HMG-CoA reductase inhibitor. Human Exposure and Toxicity: Like other HMG-CoA reductase inhibitors, lovastatin occasionally causes myopathy, manifested as muscle pain, tenderness, or weakness, with creatine kinase levels exceeding ten times the upper limit of normal. Myopathy sometimes presents as rhabdomyolysis, with or without myoglobinuria leading to acute renal failure, and rare fatalities. High plasma HMG-CoA reductase inhibitory activity levels increase the risk of myopathy. Post-marketing reports indicate rare fatal and non-fatal hepatic failure in patients taking statins, including lovastatin. Lovastatin treatment of HL-60 cells induces characteristic apoptosis in a dose-dependent manner. Animal studies: Lovastatin induced optic nerve degeneration (Waller's degeneration of retinal geniculate fibers) in a dose-dependent manner in clinically normal dogs at an initial dose of 60 mg/kg/day. Waller's degeneration of the vestibular cochlea and chromatin lysis of retinal ganglion cells were also observed in dogs treated with 180 mg/kg/day of lovastatin. Central nervous system vascular lesions characterized by perivascular hemorrhage and edema, perivascular interstitial mononuclear cell infiltration, perivascular fibrin deposition, and small vessel necrosis were observed in dogs treated with 180 mg/kg/day of lovastatin. In mice, an increased incidence of non-glandular gastric mucosal papillomas was observed after daily oral administration of lovastatin at doses of 100 or 500 mg/kg. Since the gastric glandular mucosa in these rodents was unaffected, and the human stomach is composed solely of glandular mucosa, the implications of these findings for humans remain unclear. In mice, an increased incidence of hepatocellular carcinoma and adenoma was observed after 21 months of daily oral administration of lovastatin 500 mg/kg. In dogs, drug-related testicular atrophy, decreased spermatogenesis, spermatocyte degeneration, and giant cell formation were observed starting at a daily dose of 20 mg/kg. Subcutaneous injection of the open hydroxy acid form of lovastatin into newborn rats resulted in delayed passive avoidance learning in female rats. Lovastatin has been shown to cause skeletal malformations in the offspring of pregnant mice and rats administered lovastatin at a dose of 80 mg/kg/day during pregnancy. Lovastatin has not shown mutagenicity in in vitro mammalian cell systems (rat or mouse hepatocytes, V-79 cell positive mutation studies), in vitro (Chinese hamster ovary cells) or in vivo (mouse bone marrow) chromosomal aberration studies, or in microbial systems with or without metabolic activation (Ames assay). However, some in vitro evidence suggests that inhibition of HMG-CoA reductase inhibits DNA synthesis during the S phase of the cell life cycle; this inhibition appears to be due to the consumption of mevalonate and is unrelated to its conversion to cholesterol. Lovastatin has a structure similar to HMG, which is a substituent for the endogenous substrate of HMG-CoA reductase. Lovastatin is a prodrug activated in vivo via lactone ring hydrolysis to generate an α-hydroxy acid. The hydrolyzed lactone ring mimics the tetrahedral intermediate produced by the reductase, giving the drug a 20,000-fold higher affinity for HMG-CoA reductase than its natural substrate. The bicyclic moiety of lovastatin binds to the coenzyme A moiety at the active site. Hepatotoxicity Lovastatin treatment is associated with mild, asymptomatic, and usually transient elevations in serum transaminases. In a pooled analysis of large-scale prospective surveillance studies, 3% to 5% of patients experienced ALT elevations above normal, but only 0.4% had ALT levels exceeding three times the upper limit of normal (ULN), compared to 0.1% in the placebo group. The higher the lovastatin dose, the greater the probability of these elevations; 0.1%, 0.9%, and 1.5% of patients taking 20 mg, 40 mg, and 80 mg daily, respectively, experienced elevations exceeding three times the ULN. Most elevations are self-limiting and do not require dose adjustment, but discontinuation is recommended if the elevation exceeds 10 times the ULN or persists for more than 5 times. Lovastatin is also associated with significant, clinically observable liver injury, but these cases are rare. The onset of clinical injury ranges from weeks to years. The injury pattern is usually cholestatic, but hepatocellular can also occur. Rash, fever, eosinophilia, and autoimmune features are uncommon. Liver damage usually resolves rapidly after discontinuation of lovastatin, but there have been reports of fatal acute liver failure and persistent cholestasis (Case 1). Red yeast rice, a traditional Chinese medicine used to treat hyperlipidemia, has been shown to contain monacolin K, a natural component with the same chemical structure as lovastatin, which may explain its cholesterol-lowering effects. Red yeast rice has also been associated with some cases of acute liver injury and myopathy, with symptoms similar to those associated with lovastatin. In some cases, there is cross-sensitivity to liver injury between red yeast rice products and lovastatin. Probability Score: B (May lead to clinically significant liver injury). Pregnancy and Lactation Effects ◉ Overview of Lactation Use There is currently no published information regarding the use of lovastatin during lactation. Due to concerns that lovastatin may disrupt lipid metabolism in infants, it is generally believed that lovastatin should not be used during lactation. However, some scholars have pointed out that children with homozygous familial hypercholesterolemia start taking statins from the age of 1, and that statins have low oral bioavailability and low risk to breastfed infants, especially rosuvastatin and pravastatin. [1] Until more data are available, especially for breastfed newborns or preterm infants, it is best to choose other medications. ◉ Effects on breastfed infants No relevant published information was found as of the revision date. ◉ Effects on lactation and breast milk No relevant published information was found as of the revision date. Protein binding Lovastatin and its β-hydroxy acid metabolites are highly bound to human plasma proteins (>95%), mainly due to their lipophilicity. Animal studies have shown that lovastatin can cross the blood-brain barrier and placental barrier. Toxicity Data LD50 1000 mg/kg (oral in mice) Interactions …A 60-year-old Black male developed rhabdomyolysis after 14 months of lovastatin treatment. The patient was not taking any other medications previously reported to cause this adverse reaction when used in combination with lovastatin. The adverse drug reaction causality algorithm categorized this reaction as probable or likely. Rhabdomyolysis is a rare adverse reaction to lovastatin treatment. While cases of lovastatin-induced rhabdomyolysis have been reported in combination with cyclosporine, erythromycin, gemfibrozil, or niacin, this adverse reaction can occur even without these medications. Due to a lack of information from controlled studies, caution should be exercised when using these medications in combination. Based on post-marketing surveillance, the risk of myopathy may be increased when gemfibrozil, other fibrates, and lipid-lowering doses of niacin (nicotinic acid) are used in combination with HMG-CoA reductase inhibitors, possibly because these drugs can cause myopathy when used alone. Therefore, combination therapy should be used with caution. Grapefruit juice contains one or more CYP3A4 inhibitors, which can increase the plasma concentration of drugs metabolized by CYP3A4. The effect of drinking one cup (250 ml) of grapefruit juice daily is negligible (only a 34% increase in plasma HMG-CoA reductase inhibitory activity, measured by the area under the concentration-time curve), and has no clinical significance. However, because large amounts (more than 1 liter per day) can significantly increase plasma HMG-CoA reductase inhibitory activity, grapefruit juice should be avoided while taking lovastatin. Lovastatin, like many other HMG-CoA reductase inhibitors, is a substrate of cytochrome P450 3A4 (CYP3A4). Some drugs that inhibit this metabolic pathway can increase lovastatin plasma concentrations and may increase the risk of myopathy. These medications include itraconazole, ketoconazole, posaconazole, voriconazole, macrolide antibiotics erythromycin and clarithromycin, ketolide antibiotics telithromycin, HIV protease inhibitors, bosavirin, terabhivir, antidepressants nefazodone, or products containing cobistatin. Concomitant use of these medications with lovastatin is contraindicated. If short-term use of a potent CYP3A4 inhibitor is unavoidable, lovastatin treatment should be discontinued during that period. For more complete data on lovastatin interactions (20 items total), please visit the HSDB records page. Non-human toxicity values Oral LD50 in rats > 5000 mg/kg Oral LD50 in mice > 20,000 mg/kg In vitro cytotoxicity: -Primary human hepatocytes: Lovastatin (up to 10 μM, treated for 72 hours) showed no significant cytotoxicity (cell viability >90%, MTT assay) [3] -Normal human fibroblasts (MRC-5): 50 μM lovastatin reduced cell viability by only 15%, indicating selective toxicity to cancer cells (but not liver cancer cells) [2] -In vivo safety (references [3], [4]): -Healthy volunteers (40 mg/day, 4 weeks): -5%–10% of subjects experienced mild, reversible elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (1.5–2 times higher than normal); - Rare adverse reactions: muscle pain (incidence <2%) [3][4] - Rabbits fed a high-cholesterol diet (5 mg/kg/day, 28 days): no significant changes in serum creatinine, blood urea nitrogen (BUN) or liver and kidney histopathological damage [1] - Plasma protein binding rate: - Human plasma: protein binding rate = 95%–98% (balanced dialysis, 37°C, pH 7.4) [3] |
| References | |
| Additional Infomation |
Therapeutic Uses
Cholesterol-lowering drugs; HMG-CoA reductase inhibitors /Clinical Trials/ ClinicalTrials.gov is a registry and results database that indexes human clinical studies funded by public and private institutions worldwide. The website is maintained by the National Library of Medicine (NLM) and the National Institutes of Health (NIH). Each record on ClinicalTrials.gov includes a summary of the study protocol, including: the disease or condition; the intervention (e.g., the medical product, behavior, or procedure under investigation); the title, description, and design of the study; participation requirements (eligibility criteria); the location of the study; contact information for the study location; and links to relevant information from other health websites, such as the NLM's MedlinePlus (for providing patient health information) and PubMed (for providing citations and abstracts of academic articles in the medical field). Lovastatin is indexed in the database. Lovastatin tablets (USP) are indicated for adolescents (aged 10 to 17 years, at least one year after menarche) who, despite dietary therapy, still have the following conditions, to lower total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and apolipoprotein B levels: 1. LDL-C remains above 189 mg/dL; or 2. LDL-C remains above 160 mg/dL, and: there is a family history of early-onset cardiovascular disease, or the adolescent patient has two or more other cardiovascular disease risk factors. /US product label includes/ For individuals whose risk of atherosclerotic vascular disease is significantly increased due to hypercholesterolemia, lipid-lowering therapy should be part of a multifactorial intervention. Lovastatin tablets (USP) are indicated for patients with primary hypercholesterolemia (type IIa and IIb) as adjunctive therapy to lower elevated total cholesterol (TC) and low-density lipoprotein cholesterol (LDL-C) levels. Lovastatin tablets may be used when dietary restrictions on saturated fat and cholesterol, along with other non-pharmacological treatments, are ineffective. /US product label contains/ For more complete data on the therapeutic uses of lovastatin (of 9 types), please visit the HSDB record page. Drug Warnings Like other HMG-CoA reductase inhibitors, lovastatin can occasionally cause myopathy, manifested as muscle pain, tenderness, or weakness, with creatine kinase (CK) levels exceeding ten times the upper limit of normal (ULN). Myopathy sometimes presents as rhabdomyolysis, with or without myoglobinuria leading to acute renal failure, and rare death. High levels of HMG-CoA reductase inhibitory activity in plasma increase the risk of myopathy. Lovastatin is contraindicated in pregnant women or women who may become pregnant. It should only be used when a woman of childbearing age is extremely unlikely to become pregnant and has been informed of the potential risks. If a patient becomes pregnant while taking lovastatin, the drug should be discontinued immediately, and the patient should be informed of the potential harm to the fetus. Lovastatin use in pregnant women may lower fetal mevalonic acid levels, a precursor to cholesterol biosynthesis. Atherosclerosis is a chronic process, and discontinuation of lipid-lowering drugs during pregnancy typically has minimal impact on the long-term risks associated with primary hypercholesterolemia. Therefore, women who are pregnant or may become pregnant should not use lovastatin. Lovastatin should only be used when a woman of childbearing age is highly unlikely to become pregnant and has been informed of the potential risks. Treatment should be discontinued immediately upon confirmation of pregnancy. Post-marketing reports indicate rare fatal and non-fatal liver failure in patients taking statins, including lovastatin. If severe liver injury with clinical symptoms and/or hyperbilirubinemia or jaundice occurs during lovastatin treatment, treatment should be discontinued immediately. Lovastatin should not be restarted unless another cause is identified. For more drug warnings (full version) (30 items) on lovastatin, please visit the HSDB records page. Pharmacodynamics Lovastatin is an oral lipid-lowering drug that reversibly inhibits HMG-CoA reductase. Lovastatin is used to lower plasma concentrations of total cholesterol, low-density lipoprotein cholesterol (LDL-C), apolipoprotein B (apoB), non-high-density lipoprotein cholesterol (non-HDL-C), and triglycerides (TG), while increasing high-density lipoprotein cholesterol (HDL-C) concentrations. High LDL-C, low HDL-C, and high TG concentrations in plasma are associated with an increased risk of atherosclerosis and cardiovascular disease. The ratio of total cholesterol to HDL-C is a strong predictor of coronary artery disease, and a high ratio is associated with a higher risk of disease. Elevated HDL-C levels are associated with a reduced cardiovascular risk. Lovastatin reduces the morbidity and mortality of cardiovascular disease by lowering LDL-C and TG and increasing HDL-C. Elevated cholesterol levels, especially elevated low-density lipoprotein cholesterol (LDL) levels, are an important risk factor for cardiovascular disease. Multiple landmark studies have demonstrated that using statins to lower low-density lipoprotein cholesterol (LDL-C) levels significantly reduces the risk of cardiovascular disease (CVD) and all-cause mortality. Because statins can reduce all-cause mortality, including fatal and non-fatal CVD, and decrease the need for revascularization or angioplasty after a heart attack, they are considered a cost-effective CVD treatment option. Evidence suggests that even in low-risk individuals (with a 5-year risk of a major vascular event <10%), statins can reduce the incidence of major cardiovascular events (heart attack, stroke, coronary revascularization, and coronary artery disease death) by a relative 20%-22% for every 1 mmol/L reduction in LDL-C, without significant side effects or risks. Clinical studies have shown that lovastatin can reduce LDL-C and total cholesterol by 25%-40%. The known half-maximal inhibitory dose (ICD) is 46 mcg/kg, equivalent to a reduction of approximately 30% in plasma cholesterol. Myopathy/Rhabdomyolysis: Like other HMG-CoA reductase inhibitors, lovastatin can occasionally cause myopathy, manifested as muscle pain, tenderness, or weakness, with creatine kinase (CK) levels exceeding ten times the upper limit of normal (ULN). Myopathy sometimes presents as rhabdomyolysis, with or without myoglobinuria leading to acute renal failure, and rare fatalities. The risk of myopathy is dose-related; higher plasma HMG-CoA reductase inhibitory activity levels are associated with a greater risk. In a clinical study (EXCEL), researchers closely monitored patients and ruled out drug interactions. Results showed that 1 case of myopathy occurred in 4933 patients randomly assigned to receive 20 to 40 mg lovastatin daily for 48 weeks, while 4 cases occurred in 1649 patients randomly assigned to receive 80 mg lovastatin daily. Predisposing factors for myopathy include advanced age (≥65 years), female sex, uncontrolled hypothyroidism, and renal impairment. Chinese patients may also have a higher risk of developing myopathy. In most cases, muscle symptoms and elevated creatine kinase (CK) levels resolve upon timely discontinuation of the medication. The risk of myopathy may increase if lovastatin is taken concurrently with interacting medications such as fenofibrate, niacin, gemfibrozil, cyclosporine, and potent CYP3A4 inhibitors. HMG-CoA reductase inhibitors have been reported to cause myopathy, including rhabdomyolysis, when used in combination with colchicine; therefore, caution should be exercised when taking these two medications concurrently. Real-world data from observational studies indicate that 10-15% of patients taking statins may experience muscle pain during treatment. Liver dysfunction: In early clinical trials, 1.9% of adult patients receiving lovastatin for at least one year experienced persistently elevated serum transaminases (more than 3 times the upper limit of normal). When these patients discontinue or stop taking the medication, transaminase levels typically return to pre-treatment levels slowly. Elevated transaminase levels usually occur 3 to 12 months after the start of lovastatin treatment and are not accompanied by jaundice or other clinical signs or symptoms. In the EXCEL study, the incidence of persistently elevated serum transaminase levels over 48 weeks in patients receiving lovastatin was: 0.1% in the placebo group, 0.1% in the 20 mg/day group, 0.9% in the 40 mg/day group, and 1.5% in the 80 mg/day group. However, in post-marketing experience with lovastatin, symptomatic liver disease has been reported rarely at all doses. Lovastatin is a naturally derived (from Penicillium citrinum) HMG-CoA reductase inhibitor and was the first statin approved by the FDA in 1987 for the treatment of hypercholesterolemia and the prevention of atherosclerotic cardiovascular diseases (such as myocardial infarction)[4] - Core lipid-lowering mechanism: As a prodrug, lovastatin is activated in the liver to inhibit HMG-CoA reductase, thereby blocking the synthesis of mevalonate (a key intermediate in cholesterol biosynthesis). Reduced cholesterol production can upregulate hepatic low-density lipoprotein (LDL) receptors, thereby increasing the clearance of LDL-C in the blood [1][3][4] - In addition to its lipid-lowering effect: lovastatin also has pleiotropic effects, including p53-dependent antitumor activity against liver cancer cells (supporting potential anticancer research) and regulation of blood-brain barrier permeability/leukocyte migration (suggesting its application value in autoimmune neurological diseases such as multiple sclerosis) [2][5] - Clinical limitations: low oral bioavailability, requiring co-administration with food (to improve solubility), and caution should be exercised when used in combination with CYP3A4 inhibitors (such as erythromycin) to avoid increased plasma concentrations and potential muscle toxicity [3][4] |
| Molecular Formula |
C24H36O5
|
|
|---|---|---|
| Molecular Weight |
404.54
|
|
| Exact Mass |
404.256
|
|
| CAS # |
75330-75-5
|
|
| Related CAS # |
Lovastatin (Standard);75330-75-5;Lovastatin-d9;Lovastatin-d3;1002345-93-8
|
|
| PubChem CID |
53232
|
|
| Appearance |
White to off-white solid powder
|
|
| Density |
1.1±0.1 g/cm3
|
|
| Boiling Point |
559.2±50.0 °C at 760 mmHg
|
|
| Melting Point |
175°C
|
|
| Flash Point |
185.3±23.6 °C
|
|
| Vapour Pressure |
0.0±3.4 mmHg at 25°C
|
|
| Index of Refraction |
1.532
|
|
| LogP |
4.07
|
|
| Hydrogen Bond Donor Count |
1
|
|
| Hydrogen Bond Acceptor Count |
5
|
|
| Rotatable Bond Count |
7
|
|
| Heavy Atom Count |
29
|
|
| Complexity |
666
|
|
| Defined Atom Stereocenter Count |
8
|
|
| SMILES |
CC[C@H](C)C(=O)O[C@H]1C[C@H](C=C2[C@H]1[C@H]([C@H](C=C2)C)CC[C@@H]3C[C@H](CC(=O)O3)O)C
|
|
| InChi Key |
PCZOHLXUXFIOCF-BXMDZJJMSA-N
|
|
| InChi Code |
InChI=1S/C24H36O5/c1-5-15(3)24(27)29-21-11-14(2)10-17-7-6-16(4)20(23(17)21)9-8-19-12-18(25)13-22(26)28-19/h6-7,10,14-16,18-21,23,25H,5,8-9,11-13H2,1-4H3/t14-,15-,16-,18+,19+,20-,21-,23-/m0/s1
|
|
| Chemical Name |
[(1S,3R,7S,8S,8aR)-8-[2-[(2R,4R)-4-hydroxy-6-oxooxan-2-yl]ethyl]-3,7-dimethyl-1,2,3,7,8,8a-hexahydronaphthalen-1-yl] (2S)-2-methylbutanoate
|
|
| Synonyms |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
|
|||
|---|---|---|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.18 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.18 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. View More
Solubility in Formulation 3: ≥ 2.5 mg/mL (6.18 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. Solubility in Formulation 4: 30% PEG400+0.5% Tween80+5% propylene glycol:30 mg/mL |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4719 mL | 12.3597 mL | 24.7194 mL | |
| 5 mM | 0.4944 mL | 2.4719 mL | 4.9439 mL | |
| 10 mM | 0.2472 mL | 1.2360 mL | 2.4719 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04297033 | Not yet recruiting | Drug: Lovastatin Drug: Placebo |
Cerebral Arteriovenous Malformation | Beijing Tiantan Hospital | January 1, 2021 | Phase 2 |
| NCT01527669 | Completed | Drug: LipoCol Forte capsules Drug: Lovastatin Tablet |
Healthy Subjects | National Taiwan University Hospital | February 2012 | Phase 4 |
| NCT00585052 | Terminated Has Results | Drug: Paclitaxel Drug: Lovastatin |
Ovarian Cancer | University of Iowa | August 2003 | Phase 2 |
| NCT01346670 | Completed | Drug: LipoCol and Mevacor | Healthy Volunteer | Taipei Medical University WanFang Hospital |
October 2006 | Phase 4 |
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved





























 NMR
NMR