| Size | Price | Stock | Qty |
|---|---|---|---|
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
































Purity: ≥98%
Gaboxadol ( Lu-02030, THIP, OV-101; Lu-02-030; MK-0928) is a potent GABA agonist. It is an agonist of the GABAA receptor and an antagonist of GABAC receptors (IC50 = 25 μM). Gaboxadol can also be a non-opioid analgesic and a novel type of hypnotic.
| Targets |
GABA receptor
|
|---|---|
| ln Vitro |
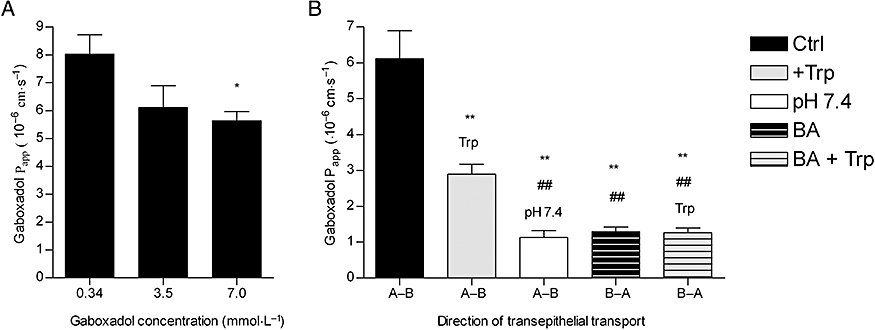
The permeability of Caco-2 cell monolayers was lowered in a dose-dependent manner by gaboxadol hydrochloride (0.34, 3.5, and 7.0 μM), with mean Papp values of 8.1 × 10-6 cm·s-1, 6.1 × 10 -1 cm·s-1 for 0.34, 3.5, and 7 μM gaboxadol, and 5.6 × 10-6 cm·s-1 for 0.34, 3.5, and 7 μM gaboxadol, respectively[3].
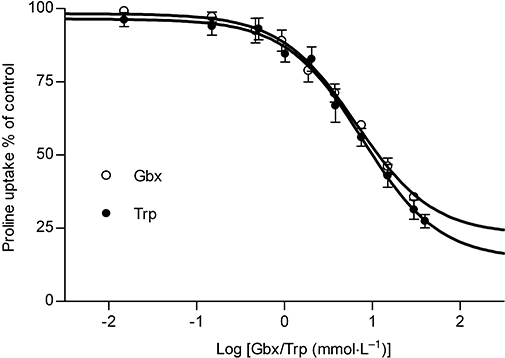
Characterization of Gaboxadol transport via hPAT1 in vitro [3] The interaction between gaboxadol and hPAT1 was investigated by measuring the apical uptake of the hPAT1 substrate proline into Caco-2 cell monolayers in the presence of increasing gaboxadol concentrations (Figure 1). Gaboxadol decreased apical proline uptake in Caco-2 cell monolayers with an estimated inhibitor affinity (Ki value) of 6.6 mmol·L−1. Similarly, the known PAT1 inhibitor, tryptophan, also decreased the apical uptake of proline with a Ki value of 7.7 mmol·L−1. The transepithelial (A-B) flux of gaboxadol transport across Caco-2 cell monolayers was investigated at three apical concentrations (0.34, 3.5 and 7.0 mmol·L−1). The mean Papp values of gaboxadol transport were 8.1 × 10−6 cm·s−1, 6.1 × 10−6 cm·s−1 and 5.6 × 10−6 cm·s−1 for 0.34, 3.5 and 7 mmol·L−1 gaboxadol, respectively (Figure 2). Thus, the gaboxadol permeability across Caco-2 cell monolayers decreased with increasing gaboxadol concentrations (P < 0.05). The gaboxadol transport across Caco-2 cell monolayers using an apical concentration of 3.5 mmol·L−1 gaboxadol was investigated in the presence of 35 mmol·L−1 tryptophan, with or without a pH gradient, and bidirectional (Figure 2). Transport of gaboxadol in the A–B direction was approximately five times higher than in the B–A direction (P < 0.005). The presence of tryptophan reduced the permeability of gaboxadol by 53% (to a Papp of 2.9 × 10−6 cm·s−1, P < 0.005). In the absence of a proton gradient across the monolayer, the permeability of gaboxadol was reduced by 82% to 1.1 × 10−6 cm·s−1 (P < 0.005). Gaboxadol permeability in the presence of tryptophan, in absence of a proton gradient, and in the B–A direction was similar to the permeability of [3H]-mannitol (Papp of 1.6 ± 0.36 × 10−6 cm·s−1). Furthermore, the presence of gaboxadol and tryptophan in the transport experiments did not change the permeability of metoprolol or mannitol transport across Caco-2 cell monolayers. The permeabilities of mannitol and metoprolol were 1.6 ± 0.36 × 10−6 cm·s−1 and 6.9 ± 0.99 × 10−6 cm·s−1, respectively. In summary, the transepithelial gaboxadol transport across Caco-2 cell monolayers was pH-dependent, could be inhibited by tryptophan and was polarized in the A-B direction. Together these observations suggest that hPAT1 mediates gaboxadol transport across the luminal membrane of human intestinal epithelial cells, and that this transport step to a large degree determines the resulting transepithelial transport of gaboxadol. Gaboxadol is a substrate for the proton-coupled amino acid transporter, hPAT1, in Caco-2 cell monolayers [3] Gaboxadol inhibited the apical uptake of the hPAT1 substrate proline in Caco-2 cell monolayers with a Ki value of 6.6 mmol·L−1. This affinity is comparable with those recently observed for other hPAT1 substrates such as GABA (3.1 mmol·L−1) and the GABA analogues muscimol (1.7 mmol·L−1) and THPO (11.3 mmol·L−1) (Larsen et al., 2008). The affinity of tryptophan was found to be quite similar to gaboxadol, that is, 7.7 mmol·L−1. Metzner et al. (2005) previously characterized tryptophan as an inhibitor of PAT1, and reported a Ki value of 4.7 mmol·L−1 for proline uptake via hPAT1 in Caco-2 cells. Considering minor differences in the proline affinities for hPAT1 between the two laboratories; Metzner et al. reports a Kt of 1.4 mmol·L−1 whereas Larsen et al. reports a Km of 3.6 mmol·L−1, respectively, the affinities of tryptophan for hPAT1 are quite comparable between the two studies (Metzner et al., 2005; Larsen et al., 2008). In accordance with results published by other groups (Thwaites et al., 1993; Metzner et al., 2005), we found that the majority of apical proline transport in Caco-2 cells was mediated by hPAT1, and no evidence of other sodium-dependent or sodium-independent transporters of proline was observed (Larsen et al., 2008). Other amino acid transporters such as the apical sodium-dependent amino acid transporters B0 (B0AT1), B0,+ (ATB0,+) and system (ASC) (ASCT2) are not likely to be involved in the transport of gaboxadol. They are all present in Caco-2 cells but their translocation of substrate is not proton-coupled. Furthermore, these transporters are characterized by higher affinity values for their substrates than observed for PAT1, for example, ASC (ASCT2), approximately 100 µmol·L−1 (Uchiyama et al., 2005); B0 (B0AT1), 500–700 µmol·L−1 (Broer et al., 2004) and B0,+ (ATB0,+), approximately 150 µmol·L−1 (Hatanaka et al., 2002). The transepithelial transport of Gaboxadol across Caco-2 cell monolayers was polarized in the apical to basolateral direction. Gaboxadol transport could be inhibited by tryptophan and was dependent on the pH of the apical donor solution. Furthermore, the Papp of gaboxadol in the apical–basolateral direction decreased with increasing gaboxadol concentration. This is consistent with transport of gaboxadol via hPAT1, and this pathway accounted for approximately 80% of the total transepithelial transport. The amino acid transport system b0,+ has been identified in the small intestine of mouse and in Caco-2 cells, where it accounts for 15–85% of the total transport of alanine and arginine respectively (Wenzel et al., 2001; Dave et al., 2004). However, tryptophan binding to system b0,+ has not been unequivocally shown (Su et al., 1992; Tate et al., 1992), and furthermore cationic amino acids, zwitterionic amino acids and cystine have µmol·L−1 affinities for system b0,+ (Palacin, 1994). Therefore, if gaboxadol is transported via system b0,+ to any significant degree, it should be evident in Caco-2 cells. In vitro the transepithelial gaboxadol transport was furthermore pH-dependent, and PAT1 is the only currently known proton-coupled amino acid transporter in the intestine. The permeability of gabapentin (also a zwitterionic γ-amino analogue) in rat small intestine was shown to be proton independent (Nguyen et al., 2007), hence different apical transport mechanisms exist for gaboxadol and gabapentin. The results indicate that gaboxadol is a substrate for hPAT1 in Caco-2 cell monolayers, and that hPAT1 mediates gaboxadol transport across the luminal membrane of the intestinal enterocytes, which appear to be important for the resulting transepithelial transport. The mechanism for gaboxadol efflux across the basolateral membrane is still unknown. In vivo absorption of Gaboxadol in dogs [3] The in vivo absorption of gaboxadol in dogs occurred rapidly following oral administration with a Tmax of approximately 0.46 h and a high bioavailability of 85%. These observations are consistent with previous studies on the oral absorption in humans showing a gaboxadol Tmax of approximately 0.5 h and a bioavailability >90% (Schultz et al., 1981; Lund et al., 2006). Once absorbed, gaboxadol is mainly excreted in the urine in the form of gaboxadol, whereas a minor fraction is excreted in the form of a glucuronic acid conjugate comprising of 2–7% in rat and mouse and 30–35% in two human subjects (Schultz et al., 1981; Lund et al., 2006; Shadle et al., 2006). Collectively, this indicates that in dogs gaboxadol is quickly and completely absorbed, probably in the proximal small intestine, with a minimal post-absorptive metabolism. |
| ln Vivo |
Gaboxadol hydrochloride (intraperitoneal injection; 0.5, 1, 1.5, 2, 3, 4, or 5 mg/kg; thrice daily; three days apart) normalizes walking distance in Fmr1 KO2 mice to 0.5 mg/kg WT activity level, in addition, the chemical had no effect on locomotor activity in Fmr1 KO2 mice [2].
Here, we sought to evaluate the potential of Gaboxadol (also called OV101 and THIP), a selective and potent agonist for delta-subunit-containing extrasynaptic GABAA receptors (dSEGA), as a therapeutic agent for FXS by assessing its ability to normalize aberrant behaviors in a relatively uncharacterized mouse model of FXS (Fmr1 KO2 mice). Four behavioral domains (hyperactivity, anxiety, aggression, and repetitive behaviors) were probed using a battery of behavioral assays. The results showed that Fmr1 KO2 mice were hyperactive, had abnormal anxiety-like behavior, were more irritable and aggressive, and had an increased frequency of repetitive behaviors compared to wild-type (WT) littermates, which are all behavioral deficits reminiscent of individuals with FXS. Treatment with gaboxadol normalized all of the aberrant behaviors observed in Fmr1 KO2 mice back to WT levels, providing evidence of its potential benefit for treating FXS. We show that the potentiation of extrasynaptic GABA receptors alone, by gaboxadol, is sufficient to normalize numerous behavioral deficits in the FXS model using endpoints that are directly translatable to the clinical presentation of FXS. Taken together, these data support the future evaluation of gaboxadol in individuals with FXS, particularly with regard to symptoms of hyperactivity, anxiety, irritability, aggression, and repetitive behaviors. [2] Gaboxadol Normalizes Hyperactivity Observed in Fmr1 KO2 Mice [2] Hyperactivity is a salient feature of human FXS (Bailey et al., 2008; Wheeler et al., 2014; Hagerman et al., 2017) and has been reliably reproduced in the previously characterized Dutch-Belgian Fmr1 KO mouse (Olmos-Serrano et al., 2010; Kazdoba et al., 2014). To test whether the Fmr1 KO2 mice showed locomotor hyperactivity and whether gaboxadol could normalize this aberrant behavior, Fmr1 KO2 mice were injected with vehicle or gaboxadol (0.5–5 mg/kg, i.p.), and WT littermates were injected with vehicle 30 min before testing in the OFT. The total distance traveled (cm) in the OFT was recorded for 30 min. The results showed that the distance traveled by Fmr1 KO2 mice was significantly increased compared to WT littermate controls (Figure 1, F(8,81) = 21.27, p < 0.0001), consistent with results from other models of FXS. Treatment with gaboxadol (0.5 mg/kg) normalized the distance traveled by Fmr1 KO2 mice to WT activity levels (Figure 1). Higher doses of gaboxadol (1–5 mg/kg, i.p.) had no effect on locomotor activity in Fmr1 KO2 mice (Figure 1). These results were not attributable to sedative effects of gaboxadol because in WT C57Bl/6 or BALB/c mice, gaboxadol doses up to 2.0 mg/kg, i.p. have no effect on locomotor activity in a 60 min OFT (data not shown), consistent with previous work showing no effect of gaboxadol on locomotion in WT mice (Olmos-Serrano et al., 2011) or rats (Silverman et al., 2016). Anxiety-Like Behaviors in Fmr1 KO2 Mice Are Normalized by Gaboxadol [2] To assess the effect of gaboxadol on anxiety-like behaviors in the Fmr1 KO2 mice, three different behavioral tests were employed: center distance traveled in the OFT, the LDT and the SAT. Increased distance traveled in the center is interpreted as decreased anxiety and takes advantage of the inherent preference of mice to remain in the perimeter when introduced to a novel environment. Fmr1 KO2 mice were injected with gaboxadol (0.5–5 mg/kg, i.p.), and WT littermates were injected with vehicle 30 min before being placed in the OFT for 30 min. The total distance traveled in the center was significantly increased in Fmr1 KO2 mice compared to WT controls (Figure 2A, F(8,81) = 21.32, p < 0.0001). Treatment with gaboxadol (0.5 mg/kg, i.p.) normalized the effect of Fmr1 KO2 on center distance traveled to levels comparable to WT controls (Figure 2A). Higher doses of gaboxadol (1–5 mg/kg) had no effect on Fmr1 KO2 mice in this test (Figure 2A). Irritability and Aggressive Behaviors in Fmr1 KO2 Mice Are Normalized by Gaboxadol [2] As with other forms of syndromic autism, a large proportion of individuals with FXS show irritability, social anxiety and aggression. These aberrant behaviors can be modeled in rodents through characterization of SIs between a test mouse and a novel cage-mate. To test the hypothesis that irritability and aggression were increased in Fmr1 KO2 mutants, we quantified instances of tail rattling, biting behavior, mounting behavior, and latency to attack. Mice were injected with vehicle or gaboxadol (0.5–5 mg/kg, i.p.) 30 min before being placed into the test cage. Tail rattling, or rapid vibrations of the tail, reflects aggressiveness and fight tendency. Fmr1 KO2 mice showed significantly increased tail rattling frequency compared to WT controls (Figure 3A, F(8,81) = 16.03, p < 0.0001). Gaboxadol (0.5, 1.5, and 5.0 mg/kg) normalized the effect in Fmr1 KO2 mice to levels comparable to WT controls (Figure 3A). Gaboxadol Normalizes Repetitive Behaviors in Fmr1 KO2 Mice [2] Perseveration and repetitive behaviors are common in individuals with FXS and are highly disruptive (Arron et al., 2011; Leekam et al., 2011; Hall et al., 2016). To test the hypothesis that such features might be observed in Fmr1 KO2 animals, we quantified circling, self-grooming, and stereotypy in WT and Fmr1 KO2 mutant mice. Counter-clockwise (CCW) revolutions were measured in the testing chamber after mice were injected with vehicle or gaboxadol (0.5–5 mg/kg, i.p.). Fmr1 KO2 mice showed significantly increased CCW revolutions during the 5 min test compared with WT controls (Figure 4A, F(8,81) = 25.46, p < 0.0001). Injection of gaboxadol (0.5, 1.0 mg/kg) into Fmr1 KO2 mice restored the number of CCW revolutions to WT levels (Figure 4A). There was no effect of genotype on clockwise circling (p = 0.386, data not shown). In vivo absorption of Gaboxadol in dogs [3] The in vivo absorption of gaboxadol in dogs occurred rapidly following oral administration with a Tmax of approximately 0.46 h and a high bioavailability of 85%. These observations are consistent with previous studies on the oral absorption in humans showing a gaboxadol Tmax of approximately 0.5 h and a bioavailability >90% (Schultz et al., 1981; Lund et al., 2006). Once absorbed, gaboxadol is mainly excreted in the urine in the form of gaboxadol, whereas a minor fraction is excreted in the form of a glucuronic acid conjugate comprising of 2–7% in rat and mouse and 30–35% in two human subjects (Schultz et al., 1981; Lund et al., 2006; Shadle et al., 2006). Collectively, this indicates that in dogs gaboxadol is quickly and completely absorbed, probably in the proximal small intestine, with a minimal post-absorptive metabolism. In vivo absorption of Gaboxadol following co-administration of tryptophan [3] Co-administration of the hPAT1 inhibitor tryptophan had a dose-dependent effect on the absorption profile of gaboxadol resulting in a decreased Cmax and an increased Tmax. A reduction in the gaboxadol absorption rate could be caused by an alteration of the gastric emptying rate. In humans, the rate of gastric emptying decreases as a function of the number of calories in an ingested meal (Calbet and MacLean, 1997; Sunesen et al., 2005), and also in dogs the composition of meals has been shown to prolong gastric emptying (Mizuta et al., 1990). To rule out that the observed effect on gaboxadol absorption was a result of altered gastric emptying, the cumulative absorption curve of paracetamol, which is often used as a marker of gastric emptying (Calbet and MacLean, 1997; Sunesen et al., 2005), was investigated in the presence of tryptophan. The gastric emptying of paracetamol was not significantly affected by high doses of tryptophan. However, a significant effect of tryptophan on the ka of gaboxadol was observed. As co-administration of tryptophan changed gaboxadol Tmax, Cmax and ka whereas the Fa, ke and the AUC were constant, the effect of tryptophan is likely to be a result of interaction between tryptophan and gaboxadol at the site of absorption, and not due to changes in gastric emptying. From other studies it is known that gaboxadol has a low degree of plasma protein binding and is not metabolized by cytochrome P-450s (Lund et al., 2006). Thus, on the basis of the findings in vitro suggesting that hPAT1 mediates the majority of the luminal gaboxadol transport in Caco-2 monolayers, it seems likely that also the in vivo observation can be explained by a PAT1-mediated gaboxadol absorption in dogs, which is reduced by the co-administration of tryptophan. The observed in vivo affinity values of tryptophan inhibition of gaboxadol intestinal transport were estimated from either the effect of tryptophan on gaboxadol Cmax or on the intestinal absorption rate constant, ka. This gave IC50 values of 10.1 and 12.6 mmol·L−1, respectively. As discussed earlier, the in vitro affinity of hPAT1 for tryptophan measured as inhibition of proline transport via hPAT1 was 7.7 mmol·L−1 in Caco-2 cells. The IC50 values are surprisingly close to each other considering that tryptophan inhibition is measured for two different compounds (proline and gaboxadol), and that the in vivo transport is composed of not only luminal transport but appearance in the systemic circulation, which encounters the transfer of gaboxadol across several membranes. hPAT1 substrates are characterized by having affinities in the millimolar range, and the transporter, which is expressed along the entire intestinal tract, has a high capacity (Chen et al., 2003). The molar dosing ratio between gaboxadol and tryptophan was up to 1:41, and as they both have comparable affinities for hPAT1, the reduced Cmax and ka may be due to the competitive interaction between gaboxadol and tryptophan at the site of absorption, that is, at the PAT1 protein in the luminal membrane of the small intestinal enterocytes. The maximal plasma concentration thus occurs with a prolonged Tmax, but due to the excessive capacity and the intestinal expression of PAT1, the absorption fraction remains unchanged as the absorption proceeds along the length of the intestine. The peak plasma concentration of gaboxadol may thus be reduced by modifying the absorption process as illustrated here, or through a more classical sustained release formulation approach as suggested earlier (Kjaer and Nielsen, 1983). |
| Cell Assay |
Protocols for cell culturing and in vitro experiments were as previously described (Larsen et al., 2008). Caco-2 cells of passages 20 through 29 were seeded onto Transwell™ inserts (1.12 cm2, 0.4 µm pore size) and experiments were conducted on day 25–28 after seeding. The apical uptake and the transepithelial transport of Gaboxadol across Caco-2 cell monolayers in apical to basolateral direction (A–B) and basolateral to apical direction (B–A) were measured in Hanks' balanced salt solution (HBSS) buffers. In all experiments, buffer applied to the basolateral side was pH 7.4. Unless otherwise stated, buffer applied in the apical chamber was adjusted to pH 6.0 after the addition of Gaboxadol hydrochloride or 35 mmol·L−1 tryptophan. The transport of 0.34, 3.5 or 7.0 mmol·L−1 Gaboxadol was investigated. These concentrations were selected based on a single bedtime oral dose of 15 mg gaboxadol to humans, which provides an approximate luminal concentration of 0.34 mmol·L−1, and the obtained gaboxadol affinity for hPAT1. Apical uptake experiments were initiated by adding fresh apical HBSS medium with 12.5 nmol·L−1 (0.5 µCi) L-(3H)proline and 0–30 mmol·L−1 gaboxadol or 0–35 mmol·L−1 tryptophan to the apical chamber. The apical uptake experiments were terminated after 5 min. Samples were analysed by scintillation counting.[3]
|
| Animal Protocol |
Animal/Disease Models: Fmr1 KO2 mice (the Fmr1 promoter and first exon are deleted, resulting in mice with missing mRNA and protein) [2]
Doses: 0.5, 1, 1.5, 2, 3, 4 or 5 mg/kg given Medication: intraperitoneal (ip) injection Experimental Results: Normalized hyperactivity was observed in Fmr1 KO2 mice. Mice in the same cage were injected with the same dose of Gaboxadol or vehicle, and mutants and controls were housed separately. All mice were group-housed in plastic cages (35 × 30 × 12 cm), five per cage, and habituated to the animal facility for at least a week before testing. The room temperature (21 ± 2°C), relative humidity (55 ± 5%), a 12 h light-dark cycle (lights on 7 am–7 pm) and air exchange (16 times per hour) were automatically controlled. All mice had ad libitum access to food and water. All testing was conducted in the light-phase by an investigator blind to genotype and drug treatment. [2] Gaboxadol Treatment and Experimental Timeline: Fmr1 KO2 mice were injected with vehicle (0.9% sterile saline) or Gaboxadol (0.5, 1, 1.5, 2, 3, 4, or 5 mg/kg, i.p.) 30 min prior to behavioral testing on each testing day, with a three-day interval between each test to avoid any cumulative effect of the drug administration. Wild-type mice injected with vehicle at the same time point were also included in all experiments. Behavioral screening of the mice (n = 10 per group) was conducted in the following order with 2–3 days between each test: Open Field Test (OFT; day 1), successive alleys (day 4), light/dark box (day 7), social tests and aggression (day 10), and self-grooming and stereotypy (day 12). [2] Absorption of Gaboxadol in dogs [3] All animal care and experimental studies were approved by the Animal Welfare Committee, appointed by the Danish Ministry of Justice, and were carried out in compliance with EC Directive 86/609/EEC, the Danish law regulating experiments on animals and NIH Guidelines for the Care and Use of Laboratory Animals. Six full-grown male beagle dogs (body weight 15.9–21.7 kg) were selected and allocated into a Roman quadrant design and assigned to receive all the six formulations of Gaboxadol hydrochloride randomly during 6 weeks. The dogs were fasted for 20–24 h before the initiation of the experiment and fed again 10 h after the administration. The gaboxadol dose was given either as an intravenous injection (1.0 mL·kg−1) or as an oral solution given by gavage (5.0 mL·kg−1) directly into the stomach using a soft tube. All dogs received 2.5 mg·kg−1 gaboxadol. In addition to gaboxadol, the oral formulations contained 0, 2.5, 10, 50 or 150 mg·kg−1 of tryptophan to ensure simultaneous co-administration of the two compounds. All solutions were adjusted to a pH of 5.2, and osmolarity was checked with a Vapro vapor pressure osmometer (model 552O, Wescor Inc., Logan, UT, USA), the intravenous solutions were adjusted to iso-osmolarity with glucose. Blood samples (2 mL) were taken from the cephalic vein by individual venepuncture and collected into Eppendorf tubes containing 200 IE heparin as an anticoagulant. Samples were collected before administration of gaboxadol and after 5, 15, 30, 60, 90 min, and 2, 3, 4, 6, 8 and 10 h after gaboxadol administration. The plasma was harvested immediately by centrifugation for 15 min at 2200 g and 4–8°C and stored at −80°C until further analysis. The animals had a 6-day washout period between treatments. Investigation of gastric emptying in dog [3] A protocol similar to the one described earlier using paracetamol as a marker was used to evaluate the influence of tryptophan on the gastric emptying rate in dogs. Six dogs (body weight 16.1–21.5 kg) were selected and randomly allocated to receive three formulations of paracetamol in a crossover study. The dogs received 50 mg·kg−1 paracetamol as an intravenous injection (1 mL·kg−1) or as an oral solution (5 mL·kg−1) containing 2.5 mg·kg−1 Gaboxadol and 0 or 150 mg·kg−1 tryptophan. Fasting of the dogs, drug administration, blood sampling and washout were done as described earlier. Analytical methods [3] Quantification of Gaboxadol in plasma and buffer: Gaboxadol was extracted from plasma and buffer samples by liquid extraction. 100 µL HBSS or plasma samples were mixed with 25 µL internal standard (d4-gaboxadol) and 25 µL purified water. Protein precipitation was carried out by addition of 400 µL cold acetonitrile. After centrifugation at 10 000 g for 15 min, 425 µL of supernatant was transferred to glass tubes and evaporated to dryness under nitrogen at 45°C. The samples were redissolved in 80 µL of methanol/acetonitrile (30:70), whirl-mixed for 10 min and centrifuged for 3 min at 3300× g. Gaboxadol was subsequently quantified by hydrophilic interaction chromatography followed by tandem mass spectrometry (MS/MS) detection using a protocol modified from Kall et al. (2007). The liquid chromatography (LC) system comprised by an Agilent 1100 series pump and degasser. An Asahipak amino column, (NH2P-50, 150 × 2 mm) from Phenomenex (Torrance, CA, USA) was used with a mobile phase of 20.0 mmol·L−1 ammonium acetate (pH 4): acetonitrile (30:70) and a flow rate of 0.2 mL·min−1. Twenty-microlitre samples were injected onto the column, which was kept at room temperature. The total run time was 10 min with the first 5 min of elution let to waste. The elution time of gaboxadol on the column was approximately 8 min. The MS/MS system used consisted of a Sciex API 4000 MS/MS detector with a Turbo Ion Spray and Turbo V source (Applied Biosystems, Foster City, CA, USA). The signals were linear between 0.5 and 2500 ng·mL−1, and the limit of quantification by this procedure was 0.5 ng·mL−1. The software was from Analyst™ (Applied Biosystems, version 4.0). |
| ADME/Pharmacokinetics |
Pharmacokinetic analysis of oral absorption of Gabosadol in dogs [3]
In beagles, plasma concentration curves were monitored over 10 hours after oral or intravenous administration of 2.5 mg·kg−1 Gabosadol (Figure 3). The bioavailability Fa of oral Gabosadol in dogs was high (over 80%) (Table 1). Oral administration of 2.5–150 mg·kg−1 tryptophan did not significantly alter the AUC of Gabosadol, and the mean relative bioavailability of each formulation ranged from 75% (10 mg·kg−1 tryptophan) to 86.1% (2.5 mg·kg−1 tryptophan). Furthermore, the co-administration of tryptophan did not alter the elimination rate constant (ke) and clearance (CL) of Gabosadol. However, after concurrent administration of 150 mg·kg⁻¹ tryptophan, the maximum plasma concentration (Cmax) of Gabosadol decreased from 2502 ng·mL⁻¹ to 1419 ng·mL⁻¹, a reduction of 57%. In addition, the time required to reach maximum plasma concentration (Tmax) was extended from 0.46 h to 1.5 h (P < 0.01). Subsequently, the Cmax values of the five dose groups were fitted to the dose-response curves (Figure 4), indicating that there is a direct interaction between the absorption of gabosadol and tryptophan concentration. The in vivo IC50 value of tryptophan to gabosadol Cmax was estimated to be 12.6 mg·kg⁻¹, equivalent to a tryptophan concentration of 12.3 mmol·L⁻¹ (uncorrected for the effect of gastrointestinal fluid dilution). Absorption rate constants of gabosadol and acetaminophen [3] As shown in the deconvolution curves in Figure 5A, the mean cumulative absorption fraction of gabosadol gradually changed with increasing tryptophan dose. Compared with gabosadol alone, the absorption of gabosadol was significantly reduced at time points of 0.5–1.25 hours in the presence of 150 mg·kg−1 tryptophan. The absorption rate of acetaminophen 60 minutes after oral administration was 91.5 ± 3.3% (Figure 5B), indicating that gastric emptying mainly occurred within the first hour after administration. Concomitant administration of 150.0 mg·kg−1 tryptophan did not significantly alter the gastric emptying rate, as the fraction of acetaminophen absorbed was not significantly different at the tested time points regardless of the presence of tryptophan. The pharmacokinetic parameters of acetaminophen in plasma, Tmax, AUC, and CL, were not significantly different from those obtained after co-administration of acetaminophen and tryptophan (results not shown). Based on the curves shown in Figure 5A, the absorption rate constant ka of gabosadol was calculated and plotted as a function of the logarithm of tryptophan dose in Figure 6A. Co-administration with tryptophan reduced the ka value of gabosadol, with an in vivo IC50 of 10.3 mg·kg−1, equivalent to an oral solution of tryptophan at a concentration of 10.1 mmol·L−1. Figure 6B shows that 150 mg·kg−1 of the PAT1 inhibitor tryptophan significantly reduced the absorption rate constant of gabosadol (P < 0.01), while it had no significant effect on the absorption rate constant of acetaminophen. |
| References |
|
| Additional Infomation |
GABA(C) receptors play a crucial role in many aspects of nervous system function, including memory, myopia, pain, and sleep, and are currently under extensive research. Evidence suggests the presence of functional GABA(C) receptors in various tissues, including the retina, hippocampus, spinal cord, superior colliculus, pituitary gland, and intestine. This review summarizes several neurochemicals that can be used to differentiate GABA(C) receptors from other major inhibitory neurotransmitter GABA receptors. Selective agonists (including (+)-cAMP and 5-methyl-IAA), competitive antagonists (such as TMPPA, (±)-cis-3-ACPBPA, and aza-THIP), positive regulators (such as allogeneolone), and negative regulators (such as epipregnenolone and lorecrix) are introduced. This article also discusses neurochemicals that may help differentiate between homologous p1 and p2 GABA(C) receptors (2-methyl-TACA and cyclothiazide). Because GABA(C) receptors are less abundant and structurally simpler than GABA(A) and GABA(B) receptors, they are highly attractive drug targets. [1] Gaboxadol at a dose of 0.5 mg/kg restored behavioral deficits in all tests in Fmr1 KO2 mice to normal. While higher doses also restored irritability and aggressive behavior, this effect was not observed in other behavioral domains assessed. One explanation for the slightly narrower therapeutic window observed here may be that previous studies have shown that both insufficient and excessive tonic inhibition lead to impaired information processing, and Gaboxadol enhances this physiological process. According to this model, the behavioral benefits of high-dose drugs are offset by drug-induced deficits unrelated to Fragile X syndrome (Duguid et al., 2012). Our results provide strong evidence for the potential benefits of Gaboxadol in reversing behaviors, aggression, and social competence associated with autism spectrum disorder. In summary, these results support the hypothesis that gabosadol's enhancement of extrasynaptic GABAA receptors may be beneficial for patients with Fragile X syndrome. In conclusion, these data support future evaluation of the use of gabosadol in patients with Fragile X syndrome, particularly in terms of symptoms such as hyperactivity, anxiety, autism spectrum disorder-related stereotyped behaviors, social skills, irritability, aggression, and cognition. [2]
Background and Objectives: Gabosadol has been developed for the treatment of chronic pain and insomnia. Clinical use of gabosadol has shown that adverse reactions appear to be related to peak serum concentrations. This study aimed to investigate the intestinal absorption mechanism of gabosadol in vitro and in vivo. Experimental Methods: In vitro transport studies were conducted using a Caco-2 cell monolayer model. In vivo pharmacokinetic studies were conducted using a beagle dog model. The dose of gabosadol was 2.5 mg·kg(-1), administered intravenously (1.0 mL·kg(-1)) or orally (5.0 mL·kg(-1)). Main Results: Gabosador is likely a substrate of the human proton-coupled amino acid transporter hPAT1 and inhibits hPAT1-mediated L-[(3)H]proline uptake in a Caco-2 cell monolayer model with an inhibition constant K(i) of 6.6 mmol·L(-1). Transepithelial transport of gabosadoc exhibits apical-to-basal polarization and is dependent on gabosadoc concentration and the pH of the apical buffer. In beagle dogs, gabosadoc was almost completely absorbed (absolute bioavailability F(a) 85.3%) with a time to peak concentration (T(max)) of 0.46 h. Oral administration of 2.5–150 mg·kg⁻¹ of the PAT1 inhibitor L-tryptophan significantly reduced the absorption rate constants k(a) and C(max) of gabosadoc and prolonged its T(max), while the area under the curve and clearance remained unchanged. Conclusions and significance: Gabosadol’s absorption across the intestinal luminal membrane may be mediated by PAT1. This study helps to reduce the absorption of gabozadol, thereby reducing peak plasma concentrations. [3] In summary, this study is the first to demonstrate that the high permeability of gabozadol across the Caco-2 cell monolayer is likely due to PAT1-mediated transluminal membrane transport, resulting in high transepithelial transport. In vitro gabozadol transport kinetics and pharmacokinetic results observed in dogs support the conclusion that PAT1-mediated gabozadol can be transported across mucosal membranes both in vitro and in vivo. Furthermore, this study demonstrates that the activity of transport proteins can be used to modulate or control the intestinal absorption of drugs. This formulation design provides a simple way to reduce peak plasma concentrations of gabozadol while maintaining high bioavailability. This may help to reduce side effects associated with high peak plasma concentrations. [3] |
| Molecular Formula |
C6H9CLN2O2
|
|---|---|
| Molecular Weight |
176.6009
|
| Exact Mass |
176.035
|
| Elemental Analysis |
C, 40.81; H, 5.14; Cl, 20.07; N, 15.86; O, 18.12
|
| CAS # |
85118-33-8
|
| Related CAS # |
THIP;64603-91-4
|
| PubChem CID |
5702253
|
| Appearance |
White to off-white solid powder
|
| Boiling Point |
295.7ºC at 760 mmHg
|
| Melting Point |
236 °C
|
| Flash Point |
132.6ºC
|
| LogP |
0.744
|
| Hydrogen Bond Donor Count |
3
|
| Hydrogen Bond Acceptor Count |
3
|
| Rotatable Bond Count |
0
|
| Heavy Atom Count |
11
|
| Complexity |
210
|
| Defined Atom Stereocenter Count |
0
|
| SMILES |
O=C1NOC2=C1CCNC2.[H]Cl
|
| InChi Key |
ZDZDSZQYRBZPNN-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C6H8N2O2.ClH/c9-6-4-1-2-7-3-5(4)10-8-6/h7H,1-3H2,(H,8,9)1H
|
| Chemical Name |
4,5,6,7-Tetrahydroisoxazolo(5,4-c)pyridin-3(2H)-one monohydrochloride
|
| Synonyms |
OV-101; OV101; Lu-02-030; MK-0928; GABOXADOL HYDROCHLORIDE; 85118-33-8; 478RVH3TVD; EINECS 285-687-7; 4,5,6,7-Tetrahydroisoxazolo[5,4-c]pyridin-3-ol hydrochloride; DTXSID90234251; GABOXADOL HYDROCHLORIDE [MI]; ISOXAZOLO(5,4-C)PYRIDIN-3(2H)-ONE, 4,5,6,7-TETRAHYDRO-, HYDROCHLORIDE (1:1); MK 0928; Lu-02030; OV 101; Lu02-030; MK0928; Lu02030; THIP
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture and light. |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
H2O : ≥ 100 mg/mL (~566.25 mM)
DMSO : ~75 mg/mL (~424.69 mM) |
|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1.47 mg/mL (8.32 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 14.7 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 1.47 mg/mL (8.32 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 14.7 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. View More
Solubility in Formulation 3: ≥ 1.47 mg/mL (8.32 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. Solubility in Formulation 4: 100 mg/mL (566.25 mM) in PBS (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 5.6625 mL | 28.3126 mL | 56.6251 mL | |
| 5 mM | 1.1325 mL | 5.6625 mL | 11.3250 mL | |
| 10 mM | 0.5663 mL | 2.8313 mL | 5.6625 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00209963 | Completed | Drug: Gaboxadol | Primary Insomnia | H. Lundbeck A/S | 2003-06 | Phase 3 |
| NCT06334419 | Recruiting | Drug: Gaboxadol Drug: Placebo |
Fragile X Syndrome | Craig Erickson | 2024-01-29 | Phase 2 |
| NCT00209846 | Completed | Drug: Gaboxadol | Primary Insomnia | H. Lundbeck A/S | 2004-06 | Phase 3 |
| NCT00209924 | Completed | Drug: Gaboxadol | Primary Insomnia | H. Lundbeck A/S | 2004-04 | Phase 3 |
| NCT02996305 | Completed | Drug: OV101 Regimen 1 Drug: OV101 regimen 2 Other: Placebo |
Angelman Syndrome | Ovid Therapeutics Inc. | 2016-01 | Phase 2 |
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved





























 COA
COA