| Size | Price | Stock | Qty |
|---|---|---|---|
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g | |||
| 10g | |||
| Other Sizes |
































Purity: ≥98%
Aripiprazole (formerly OPC-14597; OPC 14597; OPC14597; trade name Abilify) is an approved atypical antipsychotic drug that acts as a high-affinity partial agonist of 5-HT receptor. It is a powerful partial agonist at dopamine D2 and 5-HT1A receptors and an antagonist at 5-HT2A receptors, stabilizing the dopamine-serotonin system. Furthermore, aripiprazole has been reported to bind with the antagonist [3H]spiperone and the agonist [125I]7-OH-PIPAT at 0.70±0.22nM and 0.34±0.02nM, respectively.
| Targets |
5-HT1A Receptor ( Ki = 4.2 nM ); 5-HT2A Receptor; 5-HT2B Receptor; 5-HT2C Receptor; D2 Receptor; D3 Receptor; D4 Receptor
Aripiprazole (OPC-14597) is a partial agonist of dopamine D₂ receptors (rat striatal membranes, Ki = 0.34 nM) and dopamine D₃ receptors (human recombinant, Ki = 0.4 nM); it acts as a full agonist of 5-hydroxytryptamine 1A (5-HT₁A) receptors (human recombinant, Ki = 1.7 nM) and an antagonist of 5-HT₂A receptors (human cortical membranes, Ki = 3.4 nM) [1,2] - Aripiprazole (OPC-14597) has negligible affinity for dopamine D₁ receptors (Ki > 1000 nM) and muscarinic M₁ receptors (Ki > 5000 nM) in human brain membranes [3] - Aripiprazole (OPC-14597) weakly inhibits human cytochrome P450 enzyme CYP2D6 (IC₅₀ = 6.8 μM) and shows no significant inhibition of CYP3A4 (IC₅₀ > 50 μM) [4] |
|---|---|
| ln Vitro |
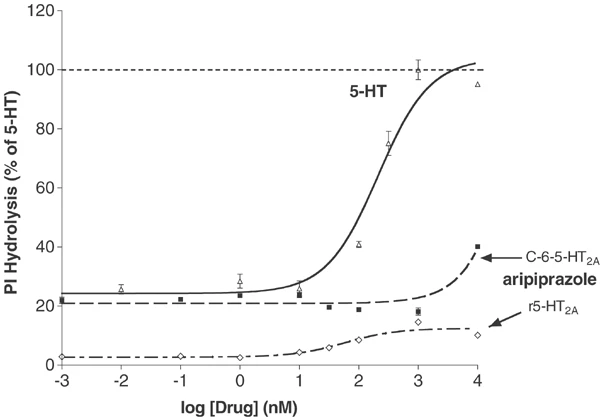
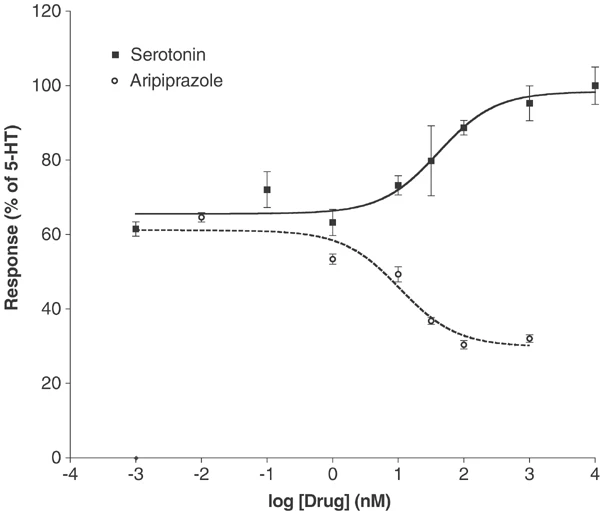
In vitro activity: Aripiprazole exhibits strong binding affinity towards both G protein-coupled and uncoupled receptor states. Aripiprazole strongly stimulates the inhibition of cAMP accumulation mediated by D2 receptors. [1] The dopamine receptors h5-HT(2B), hD(2L) and hD(3) are the ones for which aripiprazole has the highest affinity. However, it also has significant affinity (5-30 nM) for several other 5-HT receptors, including alpha(1A)-adrenergic, hH(1)-histamine, and 5-HT(1A), 5-HT(7). Other G protein-coupled receptors such as the 5-HT(1D), 5-HT(2C), alpha(1B)-, alpha(2A)-, alpha(2B)-, alpha(2C)-, beta(1)-, and beta(2)-adrenergic, and H(3)-histamine receptors are less sensitive to apipirazole (30-200 nM). Aripiprazole functions as a partial agonist at 5-HT(2A), 5-HT(2C), D(3), and D(4) receptors in addition to being an inverse agonist at 5-HT(2B) receptors. [2]
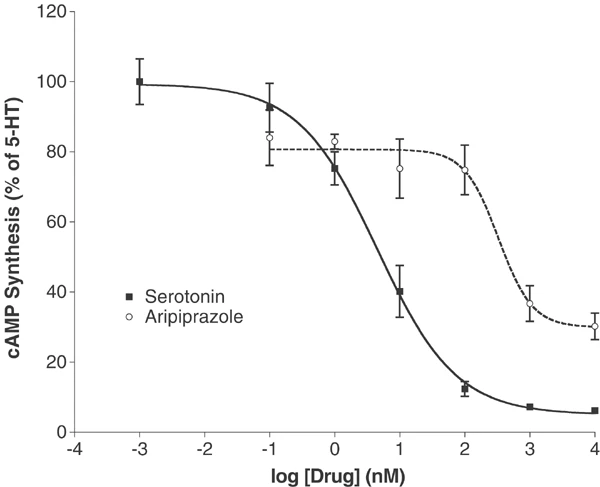
Dopamine D₂ Receptor Partial Agonism: In HEK 293 cells expressing human D₂ receptors, Aripiprazole (OPC-14597) (10⁻¹⁰ to 10⁻⁶ M) concentration-dependently stimulates cAMP production (partial agonism): maximum cAMP accumulation is 45% of the full agonist quinpirole, with an EC₅₀ of 12 nM. It antagonizes quinpirole-induced cAMP production with an IC₅₀ of 0.56 nM [3] - 5-HT₁A Receptor Agonism: In CHO cells expressing human 5-HT₁A receptors, Aripiprazole (OPC-14597) (10⁻⁹ to 10⁻⁶ M) dose-dependently inhibits forskolin-induced cAMP production (full agonism): 10⁻⁷ M reduces cAMP by 70%, with an EC₅₀ of 2.1 nM [2] - 5-HT₂A Receptor Antagonism: In rat cortical slices, Aripiprazole (OPC-14597) (10⁻⁸ to 10⁻⁶ M) concentration-dependently blocks 5-HT-induced phospholipase C activation (a marker of 5-HT₂A signaling): 10⁻⁷ M inhibits activation by 65%, with an IC₅₀ of 4.2 nM [1] - CYP2D6 Inhibition: In human liver microsomes, Aripiprazole (OPC-14597) (1–100 μM) inhibits CYP2D6-mediated dextromethorphan O-demethylation: 10 μM reduces metabolite formation by 50% (IC₅₀ = 6.8 μM), with no effect on CYP1A2 or CYP2C9 activity [4] |
| ln Vivo |
Aripiprazole reduces the levels of extracellular 5-HIAA in the striatum and medial prefrontal cortex of drug-naive rats, but not in rats that have been continuously treated with ripiprazole. [3] Rat hippocampal dopamine release is markedly increased by apiprimeze (0.1 mg/kg and 0.3 mg/kg). The release of dopamine in the medial prefrontal cortex is slightly but significantly increased by apipicazole (0.3 mg/kg), but not in the nucleus accumbens. Dopamine release in the nucleus accumbens is markedly reduced by apipicazole at doses of 3.0 mg/kg and 10 mg/kg, but not in the medical prefrontal cortex. In the medial prefrontal cortex, apipicazole (0.3 mg/kg) momentarily increases the release of dopamine induced by haloperidol (0.1 mg/kg), while inhibiting the release of dopamine in the nucleus accumbens.[4]
Rat Apomorphine-Induced Stereotypy Model: In male Sprague-Dawley rats, oral administration of Aripiprazole (OPC-14597) (1, 3, 10 mg/kg) 30 min before apomorphine (5 mg/kg, i.p.) dose-dependently reduces stereotyped behaviors (sniffing, licking): 3 mg/kg decreases total stereotypy time by 65% vs. vehicle, with no effect on locomotor activity (open-field test) [1] - Mouse Forced Swim Test (FST): In male ICR mice, oral Aripiprazole (OPC-14597) (3, 10, 30 mg/kg) 60 min before FST reduces immobility time: 10 mg/kg decreases immobility by 50% vs. vehicle, indicating antidepressant-like effects [2] - Rat Olfactory Bulbectomy (OBX) Model: In OBX rats (depression model), daily oral Aripiprazole (OPC-14597) (5 mg/kg) for 14 days reverses OBX-induced hyperactivity (reduces open-field distance by 40%) and normalizes sucrose preference (from 42% to 73%) [4] - Dog Schizophrenia-like Model: In male beagles with phencyclidine (PCP)-induced hyperlocomotion, subcutaneous Aripiprazole (OPC-14597) (0.1, 0.3 mg/kg) 15 min before PCP reduces hyperlocomotion by 70% (0.3 mg/kg) over 2 h [3] |
| Enzyme Assay |
Radioligand Binding Assays [2]
A large number of transiently and stably transfected cloned human cDNAs, obtained via the resources of the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP), were used for radioligand binding and functional assays as previously detailed (Rothman et al, 2000; Tsai et al, 2000). Conditions for radioligand binding assays, along with KD values for standard compounds, are listed in Table 1. In initial screening assays, Aripiprazole was tested at a concentration of 10 μM in quadruplicate at a large number of GPCRs, ion channels, and transporters. For molecular targets at which >50% inhibition was measured, Ki determinations were obtained using at least six concentrations of Aripiprazole; Ki values were calculated in quadruplicate using GraphPad Prism. [125I]DOI competition assays were performed as previously described (Choudhary et al, 1992) with the following changes: 12 dilutions of aAripiprazole spanning a range of 0.01–3000 nM were incubated with [125I]DOI (0.3 nM) in total volumes of 0.25 ml at 25°C for 1 h with 5–20 μg of membrane protein in binding buffer (50 mM Tris buffer, pH 7.4, 0.5 mM EDTA, 10 mM MgCl2). Membranes were harvested with a Brandel cell harvester by three ice-cold washes onto polyethyleneimine-pretreated (0.3%) Whatman GF/C filters. Radioactivity bound to filters was quantified by liquid scintillation counting. Aripiprazole is the first next-generation atypical antipsychotic with a mechanism of action that differs from currently marketed typical and atypical antipsychotics. Aripiprazole displays properties of an agonist and antagonist in animal models of dopaminergic hypoactivity and hyperactivity, respectively. This study examined the interactions of aripiprazole with a single population of human D2 receptors to clarify further its pharmacologic properties. In membranes prepared from Chinese hamster ovary cells that express recombinant D2L receptors, aripiprazole bound with high affinity to both the G protein-coupled and uncoupled states of receptors. Aripiprazole potently activated D2 receptor-mediated inhibition of cAMP accumulation. Partial receptor inactivation using the alkylating agent N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ) significantly reduced the maximum effect of aripiprazole on inhibition of cAMP accumulation. This effect was seen with concentrations of EEDQ that did not alter the maximal inhibitory effect of dopamine. Consistent with the expected effects of a partial agonist, increasing concentrations of aripiprazole blocked the action of dopamine with maximal blockade equal to the agonist effect of aripiprazole alone. The efficacy of aripiprazole relative to that of dopamine varied from 25% in cells that lacked spare receptors for dopamine to 90% in cells with receptor reserve. These results, together with previous studies demonstrating partial agonist activity at serotonin 5-hydroxytryptamine (5-HT)1A receptors and antagonist activity at 5-HT2A receptors, support the identification of aripiprazole as a dopamine-serotonin system stabilizer. The receptor activity profile may underlie the unique activity of aripiprazole in animals and its antipsychotic activity in humans. [2] Rat Striatal D₂ Receptor Binding Assay: Rat striatum was homogenized in ice-cold Tris-HCl buffer (50 mM, pH 7.4, containing 120 mM NaCl, 5 mM KCl) and centrifuged at 48,000 × g for 15 min. 50 μg of membrane protein was incubated with [³H]-spiperone (0.5 nM) and Aripiprazole (OPC-14597) (10⁻¹² to 10⁻⁶ M) at 25°C for 60 min. Non-specific binding was defined with 10 μM haloperidol. Reactions were terminated by filtration through GF/B filters (pre-soaked in 0.1% polyethyleneimine), washed 3 times, and radioactivity counted via liquid scintillation. Ki values were calculated using Cheng-Prusoff equation [1] - Human 5-HT₁A Receptor Binding Assay (CHO Cells): CHO cells expressing human 5-HT₁A receptors were homogenized in HEPES buffer (25 mM, pH 7.4, 10 mM MgCl₂) and centrifuged at 50,000 × g for 15 min. 75 μg of membrane protein was incubated with [³H]-8-OH-DPAT (0.3 nM) and Aripiprazole (10⁻¹¹ to 10⁻⁶ M) at 25°C for 90 min. Non-specific binding was determined with 10 μM methiothepin. Filtration and counting were performed as above [2] - CYP2D6 Inhibition Assay (Human Liver Microsomes): Human liver microsomes (0.5 mg protein/mL) were incubated in Tris-HCl buffer (50 mM, pH 7.4) with NADPH (1 mM), dextromethorphan (10 μM, CYP2D6 substrate), and Aripiprazole (OPC-14597) (1–100 μM) at 37°C for 30 min. Reaction was stopped with ice-cold acetonitrile, centrifuged (10,000 × g for 10 min), and supernatant analyzed via HPLC to measure dextrophan (metabolite). IC₅₀ was derived from concentration-response curves [4] |
| Cell Assay |
Effects of Aripiprazole on cAMP Production [2]
Inhibition of forskolin-stimulated cAMP production[2] Inhibition of forskolin-stimulated 3′,5′-cyclic adenosine monophosphate (cAMP) production in stable D4 and 5-HT1A receptor expressing cell lines was measured as previously reported (Lawler et al, 1999; Zhang et al, 1994). In brief, cells were grown in 24-well plates and growth media were replaced with fresh F12 medium containing 100 μM IBMX and 100 μM forskolin (all on ice) just prior to experimentation. Serial dilutions (10-fold) of Aripiprazole ranging from 0.1 to 10.000 nM were added to the cells, which were then incubated 20 min at 37°C and 5% CO2. The reaction was terminated by aspiration and the addition of 0.5 ml of ice-cold 3% trichloroacetic acid. Plates were chilled for 1 h at 4°C and spun at 1000 g for 15 min. cAMP was quantified using a competitive binding assay adapted with minor modifications (Nordstedt and Fredholm, 1990). For measurement of cAMP content, trichloroacetic acid extracts (40 μl) were added to reaction tubes containing cAMP assay buffer (100 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM EDTA). [3H]cAMP (1 nM final concentration) was added to each tube, followed by cAMP-binding proteins (approximately 100 μg of crude extract from bovine adrenal cortex in 500 μl of cAMP buffer). The reaction tubes were incubated on ice for 2 h, then harvested with a Brandel cell harvester onto Whatman GF/C filters soaked in water. Filters were allowed to dry, and bound radioactivity was quantified by liquid scintillation counting. The concentration of cAMP in each sample was estimated from a standard curve ranging from 0.1 to 100 pmol of cAMP/assay. Stimulation of cAMP production[2] Studies of the effects of serotonin and Aripiprazole at 5-HT6 and 5-HT7 receptors were carried out in stable transfectants using methods previously described (Max et al, 1995; Monsma et al, 1993; Shen et al, 1993). HEK 293 Cell D₂ Receptor Functional Assay: HEK 293 cells stably expressing human D₂ receptors were seeded in 96-well plates (1×10⁴ cells/well) and cultured in DMEM with 10% FBS for 24 h. Medium was replaced with serum-free DMEM containing Aripiprazole (OPC-14597) (10⁻¹⁰ to 10⁻⁶ M) ± quinpirole (10⁻⁷ M, full agonist). After 30 min, forskolin (10 μM) was added to stimulate cAMP. cAMP levels were measured via ELISA kit (absorbance at 450 nm) [3] - PC12 Cell Neuronal Viability Assay: PC12 cells were seeded in 24-well plates (5×10⁴ cells/well) and cultured in RPMI 1640 with 10% horse serum for 24 h. Medium was replaced with serum-free RPMI containing Aripiprazole (OPC-14597) (1, 5, 10 μM) and H₂O₂ (200 μM, oxidative stress inducer). After 24 h, cell viability was measured via MTT assay (absorbance at 570 nm): 10 μM Aripiprazole increases viability by 35% vs. H₂O₂ alone [4] |
| Animal Protocol |
0.1 mg/kg and 0.3 mg/kg
Rats Three to five days after cannulation, a dialysis probe was implanted into the medial prefrontal cortex, hippocampus or nucleus accumbens under slight anesthesia with isoflurane. Rats were then housed individually overnight in a dialysis cage. After the overnight perfusion at 0.4 μl/min of the probe, the flow was increased to 1.5 μl/min. One hour later, the dialysate samples were collected every 30 min. The perfusion medium was Dulbecco's phosphate-buffered saline solution including Ca2+ (138 mM NaCl, 8.1 mM Na2HPO4, 2.7 mM KCl, 1.5 mM KH2PO4, 0.5 mM MgCl, 1.2 mM CaCl2, pH 7.4). After stable baseline values in the dialysates were obtained, each rat received two injections, vehicle/Aripiprazole, WAY100635/aripiprazole or Aripiprazole/haloperidol. The locations of the dialysis probes were verified at the end of each experiment by brain dissection. [4] Aripiprazole was dissolved in 45% 2-hydroxypropyl-β-cyclodextrin (HBC) [4] Rat Apomorphine Stereotypy Model: Male Sprague-Dawley rats (250–300 g) were acclimated to cages for 3 days. Rats were randomized into 4 groups (n=8/group): Vehicle (0.5% methylcellulose, p.o.), Aripiprazole 1 mg/kg (p.o.), 3 mg/kg (p.o.), 10 mg/kg (p.o.). Thirty minutes post-drug, rats received apomorphine (5 mg/kg, i.p.). Stereotypy was scored every 5 min for 60 min (0=none, 3=severe), and total score calculated [1] - Mouse FST Protocol: Male ICR mice (20–22 g) were divided into 4 groups (n=10/group): Vehicle (0.5% methylcellulose, p.o.), Aripiprazole 3 mg/kg (p.o.), 10 mg/kg (p.o.), 30 mg/kg (p.o.). Sixty minutes post-gavage, mice were placed in a water cylinder (25±1°C, 15 cm depth) for 6 min. Immobility time (last 4 min) was recorded. Locomotor activity was measured 24 h later (open-field, 30 min) [2] - Rat OBX Model: Male Wistar rats (220–250 g) were anesthetized with isoflurane, and bilateral olfactory bulbs removed. Sham rats underwent surgery without bulb removal. After 14-day recovery, rats were grouped (n=7/group): Sham+Vehicle, OBX+Vehicle, OBX+Aripiprazole (5 mg/kg, p.o.). Drug was administered daily for 14 days. On day 28, open-field distance and sucrose preference (sucrose intake/total fluid) were measured [4] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion
Tablets: Aripiprazole tablets are well absorbed after administration, with peak plasma concentrations reached within 3 to 5 hours; the absolute oral bioavailability of the tablets is 87%. ABILIFY can be taken with or without food. Taking 15 mg ABILIFY tablets with a standard high-fat meal did not significantly affect the Cmax or AUC of aripiprazole or its active metabolite dehydroaripiprazole, but delayed the Tmax of aripiprazole by 3 hours and the Tmax of dehydroaripiprazole by 12 hours. Oral Solution: Aripiprazole solution is well absorbed orally. At the same dose, the plasma concentrations of aripiprazole solution are higher than those of the tablets. In a relative bioavailability study comparing the pharmacokinetics of 30 mg aripiprazole oral solution and 30 mg aripiprazole tablets in healthy subjects, the geometric mean Cmax and AUC values of the solution and tablets were 122% and 114%, respectively. Aripiprazole single-dose pharmacokinetics are linear and dose-proportional in the dose range of 5 mg to 30 mg. Sustained-release injectable suspension, administered every two months: Due to the low solubility of aripiprazole particles, the time to systemic circulation is prolonged after intramuscular injection in the buttock. The release characteristics of ABILIFY ASIMTUFII allow plasma drug concentrations to be maintained for more than 2 months after buttock injection. After multiple administrations, the median peak-to-trough ratio of aripiprazole after ABILIFY ASIMTUFII administration is 1.3, resulting in a flat plasma concentration curve. After multiple buttock administrations of 960 mg, the time to peak concentration (Tmax) ranges from 1 to 49 days. Following a single oral administration of [14C]-labeled aripiprazole, approximately 25% and 55% of the administered radioactive material, respectively, are recovered from urine and feces. Less than 1% of unmetabolized aripiprazole is excreted in urine, and approximately 18% of the oral dose is recovered in feces as unmetabolized form. After intravenous administration, aripiprazole exhibits a high steady-state volume of distribution (404 L or 4.9 L/kg), indicating its extensive extravascular distribution. The clearance of aripiprazole is estimated at 0.8 mL/min/kg. Other studies have reported clearance rates of 3297 ± 1042 mL/hr. Oral bioavailability is 87%. Aripiprazole is well absorbed and can be taken with or without food. Concomitant administration with a high-fat meal does not affect Cmax or AUC, but delays the Tmax of aripiprazole by 3 hours and the Tmax of dehydroaripiprazole by 12 hours. Time to peak concentration: Peak plasma concentration: 3 to 5 hours. The high steady-state volume of distribution of aripiprazole after intravenous administration (404 L or 4.9 L/kg) indicates its extensive extravascular distribution. At therapeutic concentrations, aripiprazole and its major metabolites bind more than 99% to serum proteins, primarily albumin. In healthy volunteers, daily administration of 0.5 to 30 mg of aripiprazole showed a dose-dependent D2 receptor occupancy, indicating that aripiprazole can cross the blood-brain barrier. For more complete data on the absorption, distribution, and excretion of aripiprazole (a total of 8 metabolites), please visit the HSDB record page. Metabolites/Metabolites Aripiprazole is primarily metabolized via three biotransformation pathways: dehydrogenation, hydroxylation, and N-dealkylation. In vitro studies have shown that CYP3A4 and CYP2D6 enzymes are responsible for the dehydrogenation and hydroxylation of aripiprazole, while N-dealkylation is catalyzed by CYP3A4. Aripiprazole is the predominantly circulating drug component. At steady state, the active metabolite dehydroaripiprazole accounts for approximately 40% of the area under the plasma concentration-time curve (AUC) of aripiprazole. Aripiprazole is extensively metabolized in the liver via dehydrogenation, hydroxylation, and N-dealkylation by cytochrome P-450 (CYP) 2D6 and 3A4 isoenzymes. The major active metabolite, dehydroaripiprazole, has a similar affinity for the D2 receptor to the parent compound, accounting for approximately 40% of the area under the plasma concentration-time curve (AUC) of aripiprazole. Steady-state plasma concentrations of both aripiprazole and dehydroaripiprazole are reached within 14 days. The activity of ABILIFY is primarily attributed to the parent drug aripiprazole, followed by its major metabolite, dehydroaripiprazole. Studies have shown that dehydroaripiprazole has a similar affinity for the D2 receptor to the parent drug, and its plasma exposure accounts for 40% of the parent drug exposure. Known metabolites of aripiprazole include dehydroaripiprazole, 4-[(2-oxo-3,4-dihydro-1H-quinoline-7-yl)oxy]butyraldehyde, 4-hydroxyaripiprazole, and 2,3-dichlorophenylpiperazine. Aripiprazole is primarily metabolized via three biotransformation pathways: dehydrogenation, hydroxylation, and N-dealkylation. Based on in vitro studies, CYP3A4 and CYP2D6 enzymes are responsible for the dehydrogenation and hydroxylation of aripiprazole, while N-dealkylation is catalyzed by CYP3A4. Aripiprazole is the predominantly circulating drug component. At steady state, the active metabolite dehydroaripiprazole accounts for approximately 40% of the plasma AUC of aripiprazole (RxList, A308). Elimination pathway: Less than 1% of unchanged aripiprazole is excreted in the urine, and approximately 18% of the oral dose is excreted unchanged in the feces. Half-life: 75–146 hours Biological half-life The mean elimination half-lives of aripiprazole and dehydroaripiprazole are approximately 75 hours and 94 hours, respectively. For individuals with poor CYP2D6 metabolism, the half-life of aripiprazole is 146 hours, and these patients should receive half the normal dose. Other studies have reported a half-life of aripiprazole of 61.03 ± 19.59 hours and a half-life of its active metabolite of 279 ± 299 hours. The mean elimination half-lives of aripiprazole and dehydroaripiprazole are approximately 75 hours and 94 hours, respectively. Oral absorption: In healthy volunteers (n=6), the Cmax of oral aripiprazole (OPC-14597) (10 mg) was 17 ng/mL (Tmax=3 h), and the absolute oral bioavailability was 87% (with no significant first-pass metabolism)[2]. Intravenous pharmacokinetics: In male Sprague-Dawley rats, the plasma clearance of intravenously administered aripiprazole (OPC-14597) (2 mg/kg) was 12 mL/min/kg, the steady-state volume of distribution (Vss) was 4.2 L/kg, and the half-life was 75 hours (due to the high tissue binding rate, the half-life is relatively long) [3] -Metabolism and excretion: Aripiprazole (OPC-14597) is mainly metabolized by CYP2D6 and secondarily by CYP3A4 to generate dehydroaripiprazole (the active ingredient, with a Ki value of 0.5 nM for D₂). In the human body, 60% of the dose is excreted in feces (metabolites) within 72 hours, 30% in urine, and <1% is excreted unchanged [4] - Tissue distribution: In male beagle dogs, the brain/plasma ratio was 4.8 2 hours after oral administration of aripiprazole (OPC-14597) (1 mg/kg), indicating that it has high blood-brain barrier penetration [3] |
| Toxicity/Toxicokinetics |
Toxicity Summary
Identification and Uses: Aripiprazole intramuscularly is used to treat acute mania associated with schizophrenia or bipolar disorder (mixed or manic) in adults, suitable for patients who require intramuscular antipsychotic medication to rapidly control behaviors that interfere with diagnosis and treatment (e.g., threatening behaviors, escalating or extremely distressing behaviors, self-destructive behaviors). Aripiprazole orally is used to treat acute irritability associated with autism. Aripiprazole orally may be used as adjunctive therapy to treat acute episodes of major depressive disorder in adults. Aripiprazole orally may be used as monotherapy or in combination with lithium or valproate to treat acute manic or mixed episodes associated with bipolar I disorder (with or without psychotic features) in adults and children aged 10–17 years. This drug may also be used orally as monotherapy or in combination with lithium or valproate for maintenance treatment of bipolar I disorder in adults and children aged 10–17 years. Aripiprazole orally is used for the acute and maintenance treatment of schizophrenia in adults and adolescents aged 13–17 years. Human Exposure and Toxicity: Antipsychotic medications, including aripiprazole, have been reported to cause a potentially fatal symptom cluster, sometimes referred to as neuroleptic malignant syndrome (NMS). Two suspected cases of NMS, both occurring during aripiprazole treatment, have been documented in the premarketing global clinical database. Clinical manifestations of NMS include high fever, muscle rigidity, altered mental status, and autonomic dysfunction (irregular pulse or blood pressure, tachycardia, excessive sweating, and arrhythmias). Other signs may include elevated creatine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure. Elderly patients with dementia-related psychosis receiving antipsychotic medication have an increased risk of death. In short-term studies for major depressive disorder (MDD) and other mental illnesses, antidepressants increased the risk of suicidal ideation and behavior (suicidal tendencies) in children, adolescents, and young adults compared to placebo. Animal Studies: In female mice, daily doses of 3 to 30 mg/kg increased the incidence of pituitary adenomas, mammary adenocarcinomas, and adenoacanthomas. Female rats were orally administered aripiprazole at doses of 2, 6, and 20 mg/kg/day from 2 weeks prior to mating until day 7 of gestation. Estrogenic cycle disturbances and increased corpus luteum were observed in all dose groups, but no impairment of fertility was observed. Increased preimplantation embryo loss was observed in the 6 mg/kg and 20 mg/kg dose groups, and decreased fetal weight was observed in the 20 mg/kg dose group. During organogenesis, pregnant rabbits were orally administered aripiprazole at doses of 10, 30, and 100 mg/kg/day. Decreased feed intake and increased abortion rate were observed in the 100 mg/kg dose group. Treatment resulted in increased fetal mortality (100 mg/kg), decreased fetal weight (30 and 100 mg/kg), increased incidence of skeletal malformations (sternal fusion in the 30 and 100 mg/kg dose groups), and minor skeletal variations (100 mg/kg). Aripiprazole and its metabolite (2,3-DCPP) exhibited chromosomal fragmentation induction in CHL cells in in vitro chromosomal aberration assays, regardless of metabolic activation. The metabolite 2,3-DCPP increased chromosomal number abnormalities in CHL cells in vitro, even without metabolic activation. A positive response was observed in a mouse micronucleus assay, but this response was confirmed to be due to a mechanism unrelated to humans. The antipsychotic activity of aripiprazole may derive from its antagonistic effects on D2 receptors in the mesolimbic pathway and 5-HT2A receptors in the frontal cortex. D2 receptor antagonism alleviates positive symptoms of schizophrenia, while 5-HT2A receptor antagonism alleviates negative symptoms. Aripiprazole has high affinity for dopamine D2 and D3 receptors, 5-HT1A and 5-HT2A receptors, and moderate affinity for dopamine D4 receptors, 5-HT2C and 5-HT7 receptors, α1-adrenergic receptors, and histamine H1 receptors. It also has moderate affinity for the 5-HT reuptake pump. Aripiprazole has no significant affinity for cholinergic muscarinic receptors. Aripiprazole functions as a partial agonist of dopamine D2 and 5-HT1A receptors, and an antagonist of 5-HT2A receptors. Interactions Substrates of hepatic microsomal enzymes: CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4; pharmacokinetic interactions are unlikely. Antihypertensive Drugs: Potential Pharmacological Interactions (Additive Antihypertensive Effects) Famotidine: Concomitant use of aripiprazole (single dose 15 mg) with a single 40 mg dose of the H2 receptor antagonist famotidine (a potent gastric acid blocker) reduces the solubility of aripiprazole, thereby decreasing its absorption rate. This results in a 37% and 21% reduction in Cmax of aripiprazole and a 13% and 15% reduction in AUC of dehydroaripiprazole, respectively. No dose adjustment of aripiprazole is required when used concurrently with famotidine. Valproic Acid: When valproic acid (500-1500 mg/day) and aripiprazole (30 mg/day) are taken concurrently at steady state, the Cmax and AUC of aripiprazole are reduced by 25%. No dose adjustment is required when aripiprazole is used concurrently with valproic acid. For more complete data on interactions of aripiprazole (12 in total), please visit the HSDB record page. Plasma protein binding: In human plasma (ultrafiltration), aripiprazole (OPC-14597) was 99% protein-bound at concentrations of 10–1000 ng/mL, and this binding was not concentration-dependent [2] -Acute toxicity: In male ICR mice, the oral LD₅₀ of aripiprazole (OPC-14597) was >2000 mg/kg; in rats, the oral LD₅₀ was >1500 mg/kg. No death or seizures were observed at a dose of 1000 mg/kg [1] -Chronic toxicity: In a 28-day rat study (dose: 10, 50, 200 mg/kg/day), the no adverse event observed dose (NOAEL) was 50 mg/kg/day. At a dose of 200 mg/kg/day, mild weight loss (5%) and an increase in AST (1.2-fold) were observed, but no histopathological changes were observed [3] - Drug interactions: In humans, co-administration of aripiprazole (OPC-14597) (10 mg, orally) with paroxetine (CYP2D6 inhibitor, 20 mg/day) increased the Cmax of aripiprazole by 2.0-fold and prolonged t₁/₂ to 100 hours [4] |
| References | |
| Additional Infomation |
Therapeutic Uses
Antipsychotic Drugs Aripiprazole (intramuscular injection) is used to treat acute agitation associated with schizophrenia or bipolar disorder (mixed or manic) in adults, for patients who are suitable for treatment with aripiprazole and require rapid control of behaviors that interfere with diagnosis and treatment (e.g., threatening behaviors, escalating or extremely distressing behaviors, self-destructive behaviors). Aripiprazole (oral) is used to treat acute irritability associated with autism. Aripiprazole (oral) is used as adjunctive therapy to treat acute episodes of major depressive disorder in adults. For more complete data on the therapeutic uses of aripiprazole (6 types), please visit the HSDB record page. Drug Warnings /Black Box Warning/ Warning: Increased mortality in patients with dementia-related psychosis. Patients with dementia-related psychosis receiving antipsychotic treatment have an increased risk of death. An analysis of 17 placebo-controlled trials (mean duration 10 weeks) showed a 1.6 to 1.7 times higher risk of death in the drug treatment group compared to the placebo group. These trials primarily involved patients taking atypical antipsychotic medications. In typical 10-week controlled trials, the mortality rate was approximately 4.5% in the drug treatment group and approximately 2.6% in the placebo group. Although the causes of death varied, most deaths appeared to be related to cardiovascular diseases (e.g., heart failure, sudden death) or infectious diseases (e.g., pneumonia). Observational studies have shown that, similar to atypical antipsychotics, treatment with conventional antipsychotics may also increase mortality. The extent to which the increased mortality observed in observational studies is attributable to antipsychotics, rather than certain patient characteristics, is currently unclear. ABILIFY (aripiperazole) is not approved for the treatment of dementia-related psychosis. /Label Contains/ /Black Box Warning/ Warning: Increased suicidal ideation and behavior. In short-term studies of major depressive disorder (MDD) and other mental illnesses, antidepressants increased the risk of suicidal ideation and behavior (suicidal tendencies) in children, adolescents, and young adults compared to placebo. Anyone considering using ABILIFY or any other antidepressant as adjunctive therapy in children, adolescents, or young adults must weigh this risk against clinical need. Short-term studies showed that antidepressants did not increase the risk of suicidal tendencies in adults 24 years of age and older compared to placebo; antidepressants reduced the risk of suicide in adults 65 years of age and older compared to placebo. Depression and certain other mental illnesses are themselves associated with an increased risk of suicide. Patients of all ages starting antidepressant therapy should be appropriately monitored and closely observed for worsening of clinical symptoms, suicidal tendencies, or abnormal changes in behavior. Family members and caregivers should be informed of the need for close monitoring and communication with the prescribing physician. ABILIFY is not approved for the treatment of childhood depression. /Included in label/ Contraindications: Known hypersensitivity to aripiprazole or any component of the formulation; such reactions range from itching/urticaria to anaphylactic shock. A 4-week, placebo-controlled clinical trial enrolled 197 children aged 10 to 17 years with bipolar disorder, demonstrating the safety and efficacy of the drug. The incidence of discontinuation due to adverse reactions was 7% in pediatric patients (aged 10 to 17 years) receiving aripiprazole and 2% in those receiving placebo. Common adverse reactions to aripiprazole in pediatric patients with bipolar disorder (incidence ≥5%, and at least twice that in the aripiprazole group compared to the placebo group) included somnolence, extrapyramidal disorder, fatigue, nausea, akathisia, blurred vision, excessive salivation, and dizziness. Although the maintenance efficacy of aripiprazole in pediatric patients has not been systematically evaluated, its maintenance efficacy can be inferred from adult data and comparisons of aripiprazole pharmacokinetic parameters in adults and pediatric patients. For more complete data on drug warnings for aripiprazole (27 in total), please visit the HSDB record page. Pharmacodynamics Aripiprazole exhibits high affinity for dopamine D2 and D3 receptors, and 5-HT1a and 5-HT2a receptors (Ki values of 0.34 nM, 0.8 nM, 1.7 nM, and 3.4 nM, respectively), and moderate affinity for dopamine D4, 5-HT2c, and 5-HT2d receptors. It also shows moderate affinity for 5-HT7, α1-adrenergic receptors, and histamine H1 receptors (Ki values of 44 nM, 15 nM, 39 nM, 57 nM, and 61 nM, respectively), and for serotonin reuptake sites (Ki = 98 nM). Aripiprazole has high affinity for dopamine D2 and D3 receptors, 5-HT1a and 5-HT2a receptors (Ki values of 0.34 nM, 0.8 nM, 1.7 nM, and 3.4 nM, respectively), and for 5-HT7, α1-adrenergic receptors, and histamine H1 receptors (Ki values of 44 nM, 15 nM, 39 nM, 57 nM, and 61 nM, respectively), and for serotonin reuptake sites (Ki = 98 nM). Aripiprazole has high affinity for serotonin D2 and D3 receptors, and for 5-HT1a and 5-HT2a receptors (Ki values of 0.34 nM, 0.8 nM, 1.7 nM, and 3.4 nM, respectively). Aripiprazole has no significant affinity for cholinergic muscarinic receptors (IC50 1000 nM). Aripiprazole (OPC-14597) is a third-generation atypical antipsychotic drug that was approved by the FDA in 2002 for the treatment of schizophrenia, bipolar disorder and major depressive disorder (adjunctive therapy)[2] - Mechanism of action: Its therapeutic effects involve dual action: 1) partial agonist of D₂ receptors (reducing positive symptoms of schizophrenia without excessively blocking dopamine); 2) 5-HT₁A receptor agonist/5-HT₂A receptor antagonist (improving negative symptoms and cognitive function)[1,3] - Clinical efficacy: In a 6-week trial of schizophrenia (n=300), aripiprazole (OPC-14597) (15 mg/day, orally) reduced PANSS scores by 40%, compared with only a 15% reduction in the placebo group. The efficacy rate (a reduction of ≥50% in PANSS score) was 62%, compared to 28% in the placebo group [2] - Safety: Aripiprazole (OPC-14597) had a lower risk of weight gain (≤2% in the 6-month trial) and a lower risk of extrapyramidal side effects (1.5% incidence of dystonia compared to 5% for haloperidol) [3] |
| Molecular Formula |
C23H27CL2N3O2
|
|
|---|---|---|
| Molecular Weight |
448.39
|
|
| Exact Mass |
447.148
|
|
| Elemental Analysis |
C, 61.61; H, 6.07; Cl, 15.81; N, 9.37; O, 7.14
|
|
| CAS # |
129722-12-9
|
|
| Related CAS # |
Aripiprazole-d8; 1089115-06-9; Aripiprazole (1,1,2,2,3,3,4,4-d8); 1089115-04-7; Aripiprazole monohydrate; 851220-85-4; 1259305-26-4 (cavoxil)
|
|
| PubChem CID |
60795
|
|
| Appearance |
White to off-white solid powder
|
|
| Density |
1.3±0.1 g/cm3
|
|
| Boiling Point |
646.2±55.0 °C at 760 mmHg
|
|
| Melting Point |
139°C
|
|
| Flash Point |
344.6±31.5 °C
|
|
| Vapour Pressure |
0.0±1.9 mmHg at 25°C
|
|
| Index of Refraction |
1.593
|
|
| LogP |
5.59
|
|
| Hydrogen Bond Donor Count |
1
|
|
| Hydrogen Bond Acceptor Count |
4
|
|
| Rotatable Bond Count |
7
|
|
| Heavy Atom Count |
30
|
|
| Complexity |
559
|
|
| Defined Atom Stereocenter Count |
0
|
|
| SMILES |
O=C1NC2=C(C=CC(OCCCCN3CCN(C4=CC=CC(Cl)=C4Cl)CC3)=C2)CC1
|
|
| InChi Key |
CEUORZQYGODEFX-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C23H27Cl2N3O2/c24-19-4-3-5-21(23(19)25)28-13-11-27(12-14-28)10-1-2-15-30-18-8-6-17-7-9-22(29)26-20(17)16-18/h3-6,8,16H,1-2,7,9-15H2,(H,26,29)
|
|
| Chemical Name |
7-[4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butoxy]-3,4-dihydro-1H-quinolin-2-one
|
|
| Synonyms |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: This product requires protection from light (avoid light exposure) during transportation and storage. |
|
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
|
|||
|---|---|---|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.58 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.58 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. View More
Solubility in Formulation 3: 2.5 mg/mL (5.58 mM) in 10% DMF 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. Solubility in Formulation 4: 2.5 mg/mL (5.58 mM) in 10% DMF 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2302 mL | 11.1510 mL | 22.3020 mL | |
| 5 mM | 0.4460 mL | 2.2302 mL | 4.4604 mL | |
| 10 mM | 0.2230 mL | 1.1151 mL | 2.2302 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
Maintenance Electroconvulsive Therapy (ECT) Versus Aripiprazole in Clozapine-resistant Schizophrenia
CTID: NCT06501339
Phase: Phase 4 Status: Not yet recruiting
Date: 2024-07-16
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved





























 COA
COA