| Size | Price | Stock | Qty |
|---|---|---|---|
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
































| ln Vitro |
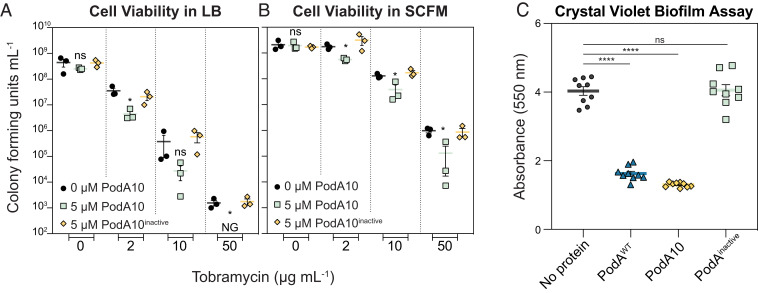
When coupled with the Mycobacterium abscessus enzyme (PodA), tobramycin (0–50 ng/mL; 24 hours) dramatically lowers M. viability of aeruginosa cells [2].
|
|---|---|
| ln Vivo |
In a mouse pneumonia model, tobramycin (50–400 mg/kg/day, intraperitoneal injection, every 4 hours) can kill germs at low doses [3].
|
| Cell Assay |
Cell Viability Assay[2]
Cell Types: Pseudomonas aeruginosa Tested Concentrations: 2,10,50 ng/mL Incubation Duration: 24 hrs (hours) Experimental Results: Cells compared to no protein or inactive Mycobacterium fortuitum enzyme (PodA) control Viability was greatly diminished, whereas PodA10 alone did not increase cell death. |
| Animal Protocol |
Animal/Disease Models: Pseudomonas aeruginosa pneumonia mouse model female, Swiss-Webster mice [3]
Doses: 50, 100. , 150, 214, and 400 mg/kg/day Dosing: intraperitonealevery 4 hrs (hrs (hours)) Experimental Results: When tobramycin was used alone, at approximately 150 mg/kg/day, the response to wild-type bacteria was Close to maximum killing effect. When used in combination with meropenem, low doses of both drugs (60 and 50 mg/kg/day for meropenem and tobramycin, respectively) produced near-maximal effects (i.e., killing of bacterial cells). Animal/Disease Models: Mice, rats, cats, and dogs for toxicological evaluation [4] Doses: 7.5, 15, 30, 120, 441,969 mg/kg Route of Administration: subcutaneous injection, intravenous (iv) (iv)injection, intramuscularinjection Experimental Results: sc LD50 values for mice and rats were 441 and 969 mg/kg respectively. Within 1 hour of treatment, rats and mice died preceded by central nervous system depression. intravenous (iv) (iv)doses of 100 mg/kg produced a moderate, transient decrease in blood pr |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion
In patients with cystic fibrosis, the variability of tobramycin concentration in sputum was greater than in serum after inhalation. Following a single inhalation of 112 mg, the serum Cmax was 1.02 ± 0.53 μg/mL with a time to peak concentration (Tmax) of 1 hour, while the sputum Cmax was 1048 ± 1080 μg/g. In contrast, after inhalation of 300 mg, the serum Cmax was 1.04 ± 0.58 μg/mL, also with a time to peak concentration of 1 hour, while the sputum Cmax was 737 ± 1028 μg/g. Systemic exposure (AUC0-12) was also similar at both doses: 4.6 ± 2.0 μg∙h/mL for the 112 mg dose and 4.8 ± 2.5 μg∙h/mL for the 300 mg dose. After four weeks of administration of tobramycin at 112 mg twice daily, the peak plasma concentration (Cmax) measured one hour after administration ranged from 1.48 ± 0.69 μg/mL to 1.99 ± 0.59 μg/mL. Tobramycin is primarily excreted unchanged in the urine. For patients with typical cystic fibrosis, the apparent volume of distribution in the central compartment of inhaled tobramycin is 85.1 L. For patients with cystic fibrosis aged 6–58 years, the apparent serum clearance of inhaled tobramycin is 14.5 L/h. Tobramycin is poorly absorbed from the gastrointestinal tract. Intramuscular absorption of tobramycin is rapid. In adults with normal renal function, after a single intramuscular dose of 1 mg/kg tobramycin, the average peak serum concentration is 4–6 μg/mL, reached within 30–90 minutes; serum concentrations decrease to 1 μg/mL or lower 6–8 hours after administration. Similar plasma drug concentrations can be achieved when the same dose is infused intravenously over 30-60 minutes. In a neonatal study, tobramycin 2 mg/kg was administered intramuscularly every 12 hours, reaching peak serum concentrations 0.5-1 hour after administration. The concentration range after the first dose was 4.9-5.2 μg/mL, and after 10-16 doses, the concentration range was 4.5-5.1 μg/mL. In neonates 2-7 days old, tobramycin was administered intravenously at a dose of 2.5 mg/kg every 12 hours. In infants weighing less than 2 kg, the steady-state peak serum concentration range was 3.5-9.9 μg/mL, and the trough concentration range was 1.1-3.6 μg/mL; in infants weighing ≥2 kg, the peak serum concentration range was 5-10.2 μg/mL, and the trough concentration range was 0.7-2 μg/mL. Due to individual differences in nebulizer performance and airway pathology, the bioavailability of tobramycin administered via nebulizer may vary. After nebulization, tobramycin is primarily concentrated within the airways; the drug does not readily cross epithelial cell membranes. Following oral inhalation of tobramycin, sputum concentrations vary considerably, but after repeated administration, the drug does not appear to accumulate in sputum. Following the first nebulization of 300 mg of commercially available tobramycin solution, the average sputum concentration at 10 minutes was 1237 μg/g (range: 35–7414 μg/g). After 20 weeks of intermittent treatment (300 mg twice daily for 28 days, followed by a 28-day break), the average sputum concentration at 10 minutes was 1154 μg/g (range: 39–8085 μg/g), and the sputum concentration at 2 hours was approximately 14% of the concentration at 10 minutes. In patients with cystic fibrosis, the mean serum tobramycin concentration 1 hour after a single inhalation of 300 mg of commercially available tobramycin solution (via nebulization) was 0.95 μg/mL; after 20 weeks of intermittent treatment (300 mg twice daily for 28 days, followed by a 28-day break), the mean serum tobramycin concentration 1.05 μg/mL was 1.05 μg/mL. For more complete data on absorption, distribution, and excretion of tobramycin (15 items in total), please visit the HSDB record page. Metabolism/Metabolites Tobramycin is hardly metabolized. Aminoglycoside antibiotics are not metabolized and are primarily excreted unchanged in the urine via glomerular filtration. /Aminoglycosides/ Biological Half-Life The apparent serum terminal half-life of tobramycin after a single inhalation of 112 mg in patients with cystic fibrosis is approximately 3 hours. Systemic clearance of tobramycin is approximately 20% higher in patients with cystic fibrosis than in those without; however, renal clearance is similar. When administered orally via nebulizer, any unabsorbed drug may be excreted primarily through sputum. ...It has been reported that in adults with normal renal function, the terminal elimination half-life exceeds 100 hours after repeated intramuscular or intravenous injections of this drug. In adults with normal renal function, the plasma elimination half-life of tobramycin after parenteral administration is typically 2–3 hours, while in adults with impaired renal function it is 50–70 hours. It has been reported that the mean elimination half-life of tobramycin in the serum of full-term infants weighing over 2.5 kg is 4.6 hours, and in the serum of infants weighing less than 1.5 kg, it is 8.7 hours. A study of newborns aged 2-7 days showed that the elimination half-life was 5.68-13.6 hours for newborns weighing less than 2 kg, and 3.54-6.73 hours for newborns weighing 2 kg or more. |
| Toxicity/Toxicokinetics |
Hepatotoxicity
Intravenous and intramuscular treatment with tobramycin typically does not cause elevated serum transaminases or bilirubin. Only sporadic case reports exist of acute liver injury with jaundice caused by aminoglycoside therapy, including tobramycin, but these are not convincing in all cases. Liver injury is usually mixed, but can also develop into cholestatic hepatitis. Onset is rapid, usually within 1 to 3 weeks, and is typically accompanied by rash, fever, and sometimes eosinophilia. Recovery usually occurs within 1 to 2 months; chronic liver injury has not been reported. Aminoglycosides were not mentioned in large case series of drug-induced liver disease and acute liver failure; therefore, liver injury caused by tobramycin, even if it occurs, is extremely rare. Pregnancy and Lactation Effects ◉ Overview of Use During Lactation Tobramycin is rarely excreted into breast milk. Neonates appear to absorb small amounts of other aminoglycoside antibiotics, but even at the typical three-times-daily dose, serum concentrations are far lower than those achieved when treating neonatal infections, making systemic effects of tobramycin unlikely. Older infants are expected to absorb even less tobramycin. Since tobramycin concentrations in breast milk fluctuate very little with multiple-dose regimens, adjusting breastfeeding and dosing times offers little benefit in reducing infant exposure. There are currently no data on once-daily dosing regimens. Monitor for potential effects on the infant's gut microbiota, such as diarrhea, candidiasis (e.g., thrush, diaper rash), or rare hematochezia, which may indicate antibiotic-associated colitis. Maternal use of ear or eye drops containing tobramycin poses little risk to breastfed infants. A working group of respiratory specialists from Europe, Australia, and New Zealand found that inhaled tobramycin is compatible with breastfeeding. ◉ Effects on breastfed infants An infant was breastfed for the fourth month postpartum (expansion of feeding not specified). At 2 months of age, the infant's mother received tobramycin treatment (150 mg three times daily) for 2 weeks due to an acute exacerbation of cystic fibrosis, concurrently with meropenem. The infant's bowel regularity remained unchanged during the mother's treatment, and renal function was normal at 6 months of age. ◉ Effects on lactation and breast milk As of the revision date, no relevant published information was found. Protein binding The binding of tobramycin to serum proteins was negligible. |
| References |
|
| Additional Infomation |
Tobramycin sulfate may cause developmental toxicity depending on state or federal labeling requirements. Tobramycin is an aminocyclic glycoside, a compound of kanamycin B lacking the 3-hydroxyl substituent on the 2,6-diaminoglycine ring. It has dual antibacterial, antimicrobial, and toxic effects. Its function is related to kanamycin B and it is the conjugate base of tobramycin (5+). Aminoglycoside antibiotics, many of which are directly derived from Streptomyces, are concentration-dependent bactericidal antibiotics with broad-spectrum antibacterial activity against both Gram-positive and Gram-negative bacteria. Inhaled tobramycin has gained attention for its use in treating chronic Pseudomonas aeruginosa infections in patients with cystic fibrosis, as Pseudomonas aeruginosa exhibits inherent resistance to many antibiotics. However, tobramycin can also be used intravenously and topically to treat a variety of infections caused by susceptible bacteria. Its use is limited in some cases by characteristic toxicities such as nephrotoxicity and ototoxicity, but it remains an important treatment option in the face of increasing resistance to first-line antibiotics such as β-lactams and cephalosporins. Tobramycin was approved by the U.S. Food and Drug Administration (FDA) in 1975 and is currently available in various dosage forms, administered via inhalation, injection, and ophthalmic administration. Tobramycin is an aminoglycoside antibiotic. It is a broad-spectrum aminoglycoside antibiotic administered parenterally and is widely used to treat moderate to severe bacterial infections caused by susceptible bacteria. Despite its widespread use, cases of liver damage associated with tobramycin are rarely observed clinically. Tobramycin has been reported to be present in Aspergillus fumigatus, Brassica napus, and other microorganisms with relevant data. Tobramycin is an aminoglycoside antibiotic derived from Streptomyces tenebrarius and possesses antibacterial activity. After entering the cell via active transport, tobramycin irreversibly binds to specific aminoglycoside receptors on the bacterial 30S ribosomal subunit, interfering with the initiation complex between messenger RNA and the 30S subunit, thereby inhibiting the initiation of protein synthesis and ultimately leading to bacterial death. Furthermore, tobramycin induces mRNA template misreading, leading to the incorporation of incorrect amino acids into the elongating polypeptide chain, thereby interfering with protein elongation. Tobramycin sulfate is the sulfate salt of tobramycin, an aminoglycoside antibiotic derived from Streptomyces tenebrarius, possessing bactericidal activity. After entering the cell via active transport, tobramycin irreversibly binds to specific aminoglycoside receptors on the bacterial 30S ribosome subunit, fixing the 30S-50S ribosome complex at the start codon (AUG), thus interfering with the initiation of protein synthesis. In addition, this drug induces mRNA template misreading, leading to: 1) dissociation of the ribosome complex, inhibiting protein elongation; or 2) incorporation of incorrect amino acids into the elongating polypeptide chain, producing abnormal or nonfunctional proteins. Cell permeability changes, ultimately leading to cell death. Tobramycin sulfate is a broad-spectrum aminoglycoside antibiotic produced by Streptomyces tenebrarius. It is effective against Gram-negative bacteria, especially Pseudomonas spp. It is a 10% component of the antibiotic complex nebramycin (NEBRAMYCIN) produced by the same bacteria.
See also: Tobramycin sulfate (in saline form); Dexamethasone; Tobramycin (component); Etapospirone; Tobramycin (component)...See more... Drug Indications Inhaled tobramycin is indicated for the treatment of cystic fibrosis caused by Pseudomonas aeruginosa, but is not recommended for children under 6 years of age, patients with a forced expiratory volume in one second (FEV1) less than 80% of the predicted value, or patients with Burkholderia cepacia. Topical tobramycin is indicated for the treatment of infections outside the eye (and adjacent structures) caused by susceptible bacteria. Tobramycin injection is indicated for the treatment of severe bacterial infections in adults and children, including sepsis (caused by Pseudomonas aeruginosa, Escherichia coli, and Klebsiella spp.), lower respiratory tract infections (caused by Pseudomonas aeruginosa, Klebsiella spp., Enterobacter spp., Serratia spp., Escherichia coli, and Staphylococcus aureus, including penicillinase-producing and non-penicillinase-producing strains), severe central nervous system infections (meningitis, caused by susceptible bacteria), intra-abdominal infections (including peritonitis, caused by Escherichia coli, Klebsiella spp., and Enterobacter spp.), and infections of the skin, bones, and skin structures. Infections caused by Pseudomonas aeruginosa, Proteus spp., Escherichia coli, Klebsiella spp., Enterobacter spp., Serratia spp., and Staphylococcus aureus, as well as complicated and recurrent urinary tract infections (caused by Pseudomonas aeruginosa, Proteus spp., Escherichia coli, Klebsiella spp., Enterobacter spp., Serratia spp., Staphylococcus aureus, Providencedum spp., and Citrobacter spp.). Aminoglycoside antibiotics, including tobramycin, are generally not used to treat uncomplicated urinary tract infections or staphylococcal infections unless a less toxic antibiotic is unavailable and the causative organism is known to be susceptible to aminoglycosides. As with all antibiotics, tobramycin use should be limited to bacterial infections known or highly suspected to be caused by susceptible organisms, and close monitoring for the development of resistance is necessary. FDA Label Vantobra® is indicated for the treatment of chronic lung infections caused by Pseudomonas aeruginosa in patients aged 6 years and older with cystic fibrosis (CF). Official guidelines for the rational use of antimicrobial agents should be consulted. Tobi Podhaler is indicated for the treatment of chronic lung infections caused by Pseudomonas aeruginosa in adults and children aged 6 years and older with cystic fibrosis. For data on different age groups, please refer to Sections 4.4 and 5.1. Official guidelines for the rational use of antibiotics should be considered. Vantobra is indicated for the treatment of chronic lung infections caused by Pseudomonas aeruginosa in patients aged 6 years and older with cystic fibrosis (CF). Official guidelines for the rational use of antibiotics should be considered. Treatment of Pseudomonas aeruginosa lung colonization in bronchiectasis patients Treatment of Pseudomonas aeruginosa lung infection/colonization in patients with cystic fibrosis Treatment of Pseudomonas aeruginosa lung infection/colonization in patients with cystic fibrosis Mechanism of Action Tobramycin is an aminoglycoside antibiotic containing a 4,6-disubstituted 2-deoxystreptamine (DOS) ring, active against a variety of Gram-negative bacteria and some Gram-positive bacteria. The mechanism of action of tobramycin is not fully elucidated, and some understanding of it relies on research findings using similar aminoglycoside antibiotics. Like other aminoglycoside antibiotics, tobramycin typically exhibits bactericidal activity, which can be both immediate and delayed, with distinct mechanisms as described below. Aminoglycoside antibiotics are polycationic at physiological pH, thus readily binding to bacterial membranes (“ion binding”); this includes binding to lipopolysaccharides and phospholipids in the outer membrane of Gram-negative bacteria, and to teichoic acid and phospholipids in the cell membrane of Gram-positive bacteria. This binding displaces divalent cations, increasing membrane permeability and allowing the aminoglycoside antibiotic to enter the cell. Further entry of aminoglycoside antibiotics into the cytoplasm (“energy-dependent phase I”) requires a proton motive force, enabling them to reach their primary intracellular target—the bacterial 30S ribosome. The mistranslated proteins (see below) generated after aminoglycoside antibiotics bind to ribosomes integrate into and disrupt the cell membrane, allowing more aminoglycoside antibiotics to enter the cell (“energy-dependent phase II”). Therefore, tobramycin and other aminoglycoside antibiotics can exert both immediate bactericidal effects by disrupting the cell membrane and delayed bactericidal effects by inhibiting protein synthesis; experimental data and mathematical models support this dual-mechanism model. Inhibition of protein synthesis is the earliest discovered mechanism of action for aminoglycoside antibiotics. Structural biology and cell biology studies have shown that aminoglycoside antibiotics bind to the 44th helix (h44) of 16S rRNA, located near the A site of the 30S ribosomal subunit, thereby altering the interaction between h44 and h45. This binding also displaces two important residues, A1492 and A1493, from h44, mimicking the normal conformational change that occurs when the codon-anticodon pair at the A site is successfully paired. In general, the binding of aminoglycoside drugs has a variety of negative effects, including inhibition of translation initiation and elongation, as well as ribosomal cycling. Recent evidence suggests that the latter effect is due to a hidden second binding site at h69 of the 50S ribosomal subunit 23S rRNA. Furthermore, aminoglycosides promote mistranslation by stabilizing a conformation that mimics correct codon-anticodon pairing; mistranslated proteins can integrate into the cell membrane, leading to the aforementioned damage. Although direct mutations in 16S rRNA are a rare resistance mechanism, post-transcriptional modifications of the 16S rRNA methyltransferase (16S-RMTases) at the N7 site of G1405 or the N1 site of A1408 are common resistance mechanisms in aminoglycoside-resistant bacteria due to the large number of copies of this gene. These mutants further support the mechanism of action of aminoglycosides. Direct modifications of aminoglycosides themselves by aminoglycoside-modifying enzymes (AMEs), such as acetylation, adenylation, and phosphorylation, are also common resistance mutations. Finally, because aminoglycosides require active transport to cross bacterial membranes, they are ineffective against obligate anaerobes. Aminoglycosides are generally effective against bacteria. Although their exact mechanisms of action are not fully elucidated, these drugs appear to inhibit protein synthesis in susceptible bacteria through irreversible binding to the 30S ribosomal subunit. /Aminoglycosides/ …Aminoglycosides are aminocyclic alcohols that kill bacteria by inhibiting protein synthesis through binding to 16S rRNA and disrupting the integrity of the bacterial cell membrane. Mechanisms of aminoglycoside resistance include: (a) inactivation of aminoglycosides through N-acetylation, adenylation, or O-phosphorylation; (b) reduction of intracellular aminoglycoside concentrations through alterations in outer membrane permeability, decreased inner membrane transport, active efflux, and drug retention; (c) alteration of the 30S ribosomal subunit target site through mutation; and (d) methylation of the aminoglycoside binding site. …/Aminoglycosides/ |
| Molecular Formula |
C18H37N5O9
|
|---|---|
| Molecular Weight |
467.51
|
| Exact Mass |
467.259
|
| Elemental Analysis |
C, 46.24; H, 7.98; N, 14.98; O, 30.80
|
| CAS # |
32986-56-4
|
| Related CAS # |
Tobramycin sulfate;49842-07-1; 32986-56-4; 79645-27-5 (sulfate deleted)
|
| PubChem CID |
36294
|
| Appearance |
White to off-white solid powder
|
| Density |
1.5±0.1 g/cm3
|
| Boiling Point |
775.4±60.0 °C at 760 mmHg
|
| Melting Point |
178ºC
|
| Flash Point |
422.8±32.9 °C
|
| Vapour Pressure |
0.0±6.0 mmHg at 25°C
|
| Index of Refraction |
1.651
|
| LogP |
-3.41
|
| Hydrogen Bond Donor Count |
10
|
| Hydrogen Bond Acceptor Count |
14
|
| Rotatable Bond Count |
6
|
| Heavy Atom Count |
32
|
| Complexity |
609
|
| Defined Atom Stereocenter Count |
14
|
| SMILES |
C1[C@@H]([C@H]([C@@H]([C@H]([C@@H]1N)O[C@@H]2[C@@H]([C@H]([C@@H]([C@H](O2)CO)O)N)O)O)O[C@@H]3[C@@H](C[C@@H]([C@H](O3)CN)O)N)N
|
| InChi Key |
NLVFBUXFDBBNBW-PBSUHMDJSA-N
|
| InChi Code |
InChI=1S/C18H37N5O9/c19-3-9-8(25)2-7(22)17(29-9)31-15-5(20)1-6(21)16(14(15)28)32-18-13(27)11(23)12(26)10(4-24)30-18/h5-18,24-28H,1-4,19-23H2/t5-,6+,7+,8-,9+,10+,11-,12+,13+,14-,15+,16-,17+,18+/m0/s1
|
| Chemical Name |
(2S,3R,4S,5S,6R)-4-amino-2-[(1S,2S,3R,4S,6R)-4,6-diamino-3-[(2R,3R,5S,6R)-3-amino-6-(aminomethyl)-5-hydroxyoxan-2-yl]oxy-2-hydroxycyclohexyl]oxy-6-(hydroxymethyl)oxane-3,5-diol
|
| Synonyms |
tobramycin; 32986-56-4; Nebramycin VI; Nebramycin 6; Nebramycin factor 6;
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
H2O : ≥ 100 mg/mL (~213.90 mM)
DMSO : ~2 mg/mL (~4.28 mM) |
|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: 100 mg/mL (213.90 mM) in PBS (add these co-solvents sequentially from left to right, and one by one), clear solution; with sonication.
(Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1390 mL | 10.6950 mL | 21.3899 mL | |
| 5 mM | 0.4278 mL | 2.1390 mL | 4.2780 mL | |
| 10 mM | 0.2139 mL | 1.0695 mL | 2.1390 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved