| Size | Price | Stock | Qty |
|---|---|---|---|
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
































Purity: ≥98%; ≥800ug/mg
| Targets |
Antibiotic
|
|---|---|
| ln Vitro |
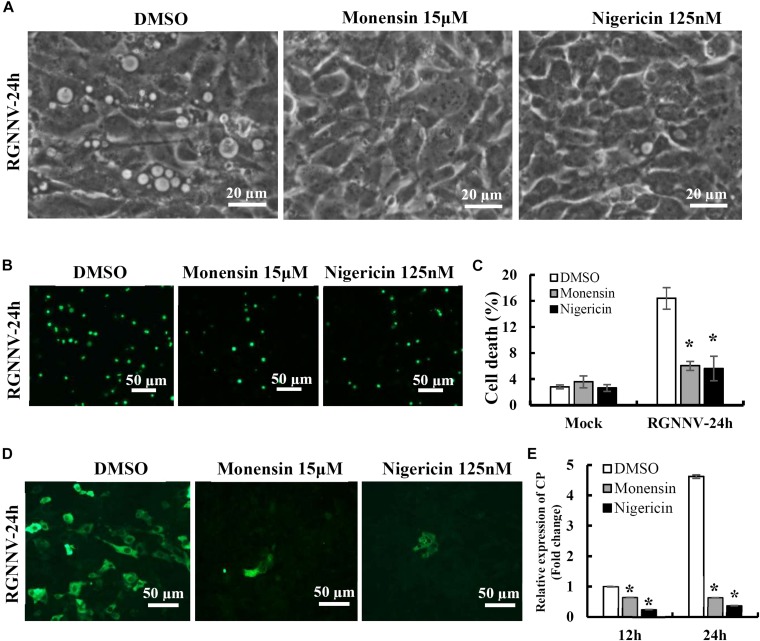
Streptomyces cinnamonensis produces the antibiotic monensin sodium salt, which causes cell death. Cells treated by default showed 2.5% sterility; 48 hours of treatment with 1 μM monensin sodium salt led to 4.5% cell death; in contrast, 48 hours of treatment with 5 μM monensin sodium salt produced a higher percentage of cell disinfection (16.4%). When monensin sodium salt was depleted at 1 or 5 μM for 24 hours, and then treated at 10 μM for 24 hours, there was a notable rise in cell engraftment events as compared to when monensin sodium salt or erlotinib was treated (14.6% and 38.7%, respectively). The combination of 10 μM erlotinib and 5 μM sodium monensin salt exhibited the largest proportion of cellular artifacts (38.7%) [1].
|
| ln Vivo |
Monensin sodium-treated Apc+/Min mice showed a substantial (P=0.0144) decrease in the mean size of lesions when compared to control animals (mean 0.199 mm2 and 0.299 mm2). However, there was no significant change in the number of tumors. One animal's estimated total tumor area decreased (mean 10.16 mm2 vs. 16.46 mm2; P=0.0125) among those receiving monensin sodium. The amount of South African cells and cells expressing the p21 cell cycle in the tumor growth surface area increased after treatment with monensin sodium salt. In healthy sections of the mucosa, there were no alterations in cell proliferation, necrosis, or tissue structure following exposure to monensin sodium salt [2].
|
| Cell Assay |
Targeting the EGFR, with inhibitors such as erlotinib, represents a promising therapeutic option in advanced head and neck squamous cell carcinomas (HNSCC). However, they lack significant efficacy as single agents. Recently, we identified the ability of statins to induce synergistic cytotoxicity in HNSCC cells through targeting the activation and trafficking of the EGFR. However, in a phase I trial of rosuvastatin and erlotinib, statin-induced muscle pathology limited the usefulness of this approach. To overcome these toxicity limitations, we sought to uncover other potential combinations using a 1,200 compound screen of FDA-approved drugs. We identified monensin, a coccidial antibiotic, as synergistically enhancing the cytotoxicity of erlotinib in two cell line models of HNSCC, SCC9 and SCC25. Monensin treatment mimicked the inhibitory effects of statins on EGFR activation and downstream signaling. RNA-seq analysis of monensin-treated SCC25 cells demonstrated a wide array of cholesterol and lipid synthesis genes upregulated by this treatment similar to statin treatment. However, this pattern was not recapitulated in SCC9 cells as monensin specifically induced the expression of activation of transcription factor (ATF) 3, a key regulator of statin-induced apoptosis. This differential response was also demonstrated in monensin-treated ex vivo surgical tissues in which HMG-CoA reductase expression and ATF3 were either not induced, induced singly, or both induced together in a cohort of 10 patient samples, including four HNSCC. These results suggest the potential clinical utility of combining monensin with erlotinib in patients with HNSCC[1].
|
| Animal Protocol |
The Wnt signaling pathway is required during embryonic development and for the maintenance of homeostasis in adult tissues. However, aberrant activation of the pathway is implicated in a number of human disorders, including cancer of the gastrointestinal tract, breast, liver, melanoma, and hematologic malignancies. In this study, we identified monensin, a polyether ionophore antibiotic, as a potent inhibitor of Wnt signaling. The inhibitory effect of monensin on the Wnt/β-catenin signaling cascade was observed in mammalian cells stimulated with Wnt ligands, glycogen synthase kinase-3 inhibitors, and in cells transfected with β-catenin expression constructs. Furthermore, monensin suppressed the Wnt-dependent tail fin regeneration in zebrafish and Wnt- or β-catenin-induced formation of secondary body axis in Xenopus embryos. In Wnt3a-activated HEK293 cells, monensin blocked the phoshorylation of Wnt coreceptor low-density lipoprotein receptor related protein 6 and promoted its degradation. In human colorectal carcinoma cells displaying deregulated Wnt signaling, monensin reduced the intracellular levels of β-catenin. The reduction attenuated the expression of Wnt signaling target genes such as cyclin D1 and SP5 and decreased the cell proliferation rate. In multiple intestinal neoplasia (Min) mice, daily administration of monensin suppressed progression of the intestinal tumors without any sign of toxicity on normal mucosa. Our data suggest monensin as a prospective anticancer drug for therapy of neoplasia with deregulated Wnt signaling[2].
|
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion
This study determined the pharmacokinetics of monensin in broilers, including half-life, apparent volume of distribution, systemic clearance, systemic bioavailability, and tissue residues. The drug was administered as a single dose of 40 mg/kg body weight via crop injection and intravenous injection. Following intravenous injection, the pharmacokinetics of monensin followed a two-compartment open model, with an absorption half-life of 0.59 hours, a volume of distribution of 4.11 L/kg, and a systemic clearance of 28.36 mL/kg/min. Peak serum monensin concentrations were reached 0.5 hours after crop injection, with an absorption half-life of 0.27 hours and an elimination half-life of 2.11 hours. Systemic bioavailability after crop injection was 65.1%. The in vitro calculated serum protein binding rate of monensin was 22.8%. Following a single intracrocal injection of pure monensin (40 mg/kg body weight), the concentrations of monensin in chicken serum and tissues were higher than those after two weeks of feeding with monensin-added premix (120 mg/kg). Monensin residues were detected in test tissues collected at 2, 4, 6, and 8 hours after oral administration. The highest concentration was found in the liver. Furthermore, monensin residues were detected only in the liver, kidneys, and fat 24 hours after the last oral administration. After 48 hours, no monensin residues were detected in any tissues other than the liver, and the residues in the liver were completely cleared within 72 hours. Six chickens were fed (3)H-monensin sodium at a dose of 121 mg/kg for two consecutive days. Only 52-73% of the radioactivity was recovered. Of this, 97% was present in the feces. The reason for the poor radioactivity balance is unknown. /Monensin Sodium/ Broiler chickens were fed (14)C-monensin sodium at a concentration of 120 mg/kg for 4 consecutive days (two males and three females) or 6 consecutive days (three males and three females). Radioactivity was detected in the liver, kidneys, fat, and skin 6 hours after the treated feed was discontinued, with the highest radioactivity found in the liver (0.5 mg/kg liver). No radioactivity was detected in muscle tissue. /Monensin Sodium/ Ten male White Leghorn roosters and two White Leghorn mother roosters were given a single oral dose of gelatin capsules containing (14)C-monensin (dose range: 2.6-100 mg). Some chickens underwent colostomy, while the rest had bile ducts inserted. The absorption rate of ingested (14)C-monensin in chickens ranged from 11% to 31%. The primary route of excretion was feces, with small amounts excreted through urine and respiration. For more complete data on the absorption, distribution, and excretion of monensin (21 items in total), please visit the HSDB record page. Metabolism/Metabolites This study investigated the oxidative metabolism of monensin in the liver microsomes of horses, pigs, broilers, cattle, and rats. Monensin is an ion-carrier antibiotic widely used in veterinary practice as an anticoccidial agent and growth promoter. By measuring the amount of formaldehyde released, the rate of monensin O-demethylation was found to be almost on the same order of magnitude across all species. However, the total monensin metabolite rate, estimated by substrate disappearance rate determined by high-performance liquid chromatography (HPLC), was highest in cattle, intermediate in rats, chicks, and pigs, and lowest in horses. When expressed as turnovers (nanomoles of monensin metabolized per minute / nanomoles of cytochrome P450⁻¹), catalytic efficiency (chicks >> cattle >> pigs ≈ rats > horses) was found to be negatively correlated with known differences in the toxicity of this ion-carrier among different species. The oral LD50 of this ionocarrier was 2-3 mg/kg body weight in horses, 50-80 mg/kg body weight in cattle, and 200 mg/kg body weight in chicks. Chicken and bovine microsomes exhibited the highest catalytic efficiency for both P450 3A-dependent substrates (erythromycin and triacetylpyridinium chloride), and also showed the highest levels of immunodetectable proteins that cross-reacted with anti-rat P450 3A1/2 antibodies. … Compared to untreated rats, monensin O-demethylation was significantly higher in phenobarbital-treated rat microsomes, and this O-demethylation was dependent on reduced nicotinamide adenine dinucleotide phosphate (NADPH), suggesting that monensin is a substrate of cytochrome P450 (CYP) enzymes. The oxidative metabolism of monensin appears to occur at least partially via CYP3A, as treatment of rat liver microsomes with a chemical inducer of CYP3A significantly increased O-demethylation of monensin. Some studies speculate that competition between monensin and other CYP3A substrates may explain the accidental poisoning events that have occurred in various livestock after simultaneous administration of monensin and other chemotherapeutic drugs, as monensin metabolism is significantly reduced in rats when other CYP3A substrates are present. Monensin metabolites are primarily derived from O-demethylation of the methoxy group and/or hydroxylation at multiple sites on the ion carrier backbone. Although sufficient monensin metabolites have been obtained for activity testing, four metabolites produced by rat liver microsomes (including O-demethylmonensin, a byproduct of monensin production) have been tested. Results showed that these metabolites exhibited antibacterial, anticoccal, cytotoxic, cardiotonic, and ion carrier activities at least 10 to 20 times lower than the parent compound, indicating that metabolism eliminates most of the biological activity of monensin. Monensin is extensively metabolized in the liver, producing more than 50 different metabolites, which have been detected in the liver, bile, and feces of chickens, cattle, rats, pigs, dogs, turkeys, sheep, and horses. In most species (chickens, mice, dogs, turkeys, and pigs), less than 10% of monensin is excreted as the parent compound, while a study of calves showed that 50-68% of the 14C detected in feces was unmetabolized monensin. This difference in the amount of metabolized monensin may be due to different species having different absorption rates of the molecule. The total metabolic rate of monensin in microsomes was estimated by determining the substrate disappearance rate using high-performance liquid chromatography (HPLC), with cattle showing the highest rate, followed by mice, chickens, and pigs, and horses showing the lowest. Metabolite patterns in laboratory and non-laboratory animals were qualitatively similar, but quantitative differences existed. No single metabolite dominated the metabolic profile. The metabolism of sodium monensin in human liver microsomes has been compared with that in horses and dogs. Mixed human microsomal samples from multiple donors (males and females, Caucasian, Hispanic, and African American, aged 15–66 years), mixed canine microsomal samples, and equine microsomal samples from a single donor were incubated with monensin at concentrations of 0.5, 1, and 10 μg/mL in the presence or absence of NADPH. Metabolite profiles were analyzed using liquid chromatography-mass spectrometry (LC-MS) at 0, 5, 10, 20, 40, and 60 minutes. Monensin is metabolized kinetically in all species and is extensively metabolized (93–99% metabolized within 60 minutes). The metabolic turnover of monensin in humans is similar to that in dogs, while the metabolic turnover in horses is only 10% of that in dogs and humans. Biological Half-Life This study determined the pharmacokinetics of monensin in broilers, including half-life, apparent volume of distribution, systemic clearance, systemic bioavailability, and tissue residues. The drug was administered in a single dose of 40 mg/kg body weight via both intravesical and intravenous injection. Following intravenous injection, the pharmacokinetics of monensin followed a two-compartment open model, with an absorption half-life of 0.59 hours… After intravesical injection, peak serum monensin concentrations were reached 0.5 hours, with an absorption half-life of 0.27 hours and an elimination half-life of 2.11 hours… |
| Toxicity/Toxicokinetics |
Toxicity Overview
Identification and Uses: Monensin is a polyether carboxylic acid ionocarboxylic acid antibiotic. Monensin is a mixture of four analogs, A, B, C, and D, with monensin A being the major component (98%). Depending on the purification method, monensin can exist in mycelial, crystalline, and recrystallized forms. It is used to treat coccidiosis in poultry (chickens, turkeys, and quail) and ruminants (cattle, sheep, and goats). Monensin is also used to control ketosis and bloating in cattle and as a growth promoter feed additive for cattle and sheep. Monensin is primarily effective against Gram-positive bacteria. Human Exposure and Toxicity: A 17-year-old boy died after ingesting monensin sodium for 11 days, developing myoglobinuria, renal failure, and death. In another case, a patient ingested monensin at three times the lethal dose for cattle and presented with clinical manifestations similar to those reported in veterinary medicine. The patient initially developed extremely severe rhabdomyolysis, which subsequently progressed to acute renal failure, heart failure, and ultimately death. The main lesions observed during autopsy included extensive skeletal muscle necrosis, myocardial complement deposition, pulmonary edema, and acute renal tubular injury. Animal experiments: Acute toxicity tests were conducted on adult rhesus monkeys. Pairs of monkeys were administered a single dose of monensin at 20, 40, or 60 mg/kg body weight via gavage and monitored for 7 days. All animals survived and developed diarrhea within 24 hours of administration. Adult goats were administered monensin sodium via gavage for five consecutive days at 13.5 mg/kg. Monensin exposure caused diarrhea, tachycardia, and decreased rumen motility and body temperature. In an inhalation exposure study, rats were exposed to normal air or air containing an average concentration of 79 mg/m³ of particulate mycelial monensin sodium for 2 weeks (1 hour daily, 5 days a week). Nine out of ten treated female rats developed anorexia and weight loss in the second week of the study. Two male and two female rats developed mild focal skeletal myositis, while the control group did not. Multifocal cardiomyopathy was observed in male rats treated with monensin. In a subchronic study, male and female mice were fed diets containing 0, 37.5, 75, 150, or 300 mg/kg of mycelial monensin sodium, respectively, for 3 months. Body weight gain decreased in a dose-dependent manner in all dose groups. At the end of the study, female and male rats experienced a 27% and 21% decrease in body weight, respectively, in the lowest dose group, while both male and female rats experienced a 99% decrease in body weight in the highest dose group. In a chronic toxicity study, male and female rats were fed diets containing 25, 56, or 125 mg/kg of crystalline monensin sodium, respectively, while the control group was fed a normal diet for 2 years. Animals fed the 125 mg/kg monensin diet showed significantly reduced body weight and weight gain, while rats in the medium dose group experienced a transient decrease in body weight and weight gain during the first 4 months. Benign and malignant tumors were observed in both treated and untreated animals, but there was no correlation between monensin administration and tumor type or severity. Monensin is toxic to horses. Clinical signs included tachycardia and arrhythmias, groaning, ataxia, sweating, recumbency, and paddling movements of the limbs before death. Post-mortem findings were concentrated in skeletal and cardiac muscles. This study investigated the effects of developmental exposure to monensin in rats. Female rats were administered monensin at concentrations of 0, 100, or 300 mg/kg until reaching a pre-mating weight of 185 g, with continued administration during gestation and lactation. Female rats in the highest-dose group experienced significant weight loss 8 days after administration. Both male and female pups in the highest-dose group showed weight loss from day 10 to day 21 postnatally. Male pups in the low-dose group showed weight loss only on day 21 postnatally. No external malformations were observed in any pups. Furthermore, this study investigated the effects of monensin (a potent Golgi apparatus disruptor) on male fertility. Male rats were administered monensin at doses of 2.5, 5, and 10 mg/kg body weight. Animals were sacrificed 67 days after treatment. Electron microscopic observations, such as cell membrane rupture, swelling, and Golgi apparatus disintegration, strongly suggested that monensin interfered with Golgi apparatus function in spermatogenic cells. Sperm count and motility data, fertility studies, and final litter size further demonstrated that monensin has an antifertility effect in male rats. Genotoxicity tests were negative. Interactions This study evaluated the combined effects of monensin (150 mg/kg) and growth promoters (GPs) zinc bacitracin (BAC, 50 mg/kg), virginiamycin (VIR, 25 mg/kg), and avopacin (AVO, 20 mg/kg) on broiler growth performance, dietary nutrient utilization, defecation and eviscerated carcass yield (DEC), and organ size in male broilers aged 7 to 28 days. The effects of growth promoters on broiler growth and carcass performance were also determined when broilers were fed a diet without monensin before 49 days of age. Results showed that monensin significantly (P < 0.05) reduced feed intake, weight gain, and feed conversion ratio in broilers aged 7 to 28 days. None of the growth promoters could offset these effects. However, AVO slightly improved these symptoms. AVO significantly increased feed intake from 7 to 28 days of age and improved weight gain and feed conversion ratio, but this was not observed from 28 to 49 days of age or from 7 to 49 days of age. VIR and BAC did not affect production performance in either of these age groups. Monensin did not affect the utilization of dietary dry matter, fat, or energy, but significantly reduced nitrogen utilization. AVO improved nitrogen and fat utilization and increased dietary AME(n) content. VIR also increased AME(n). The utilization of these nutrients was not affected by the interaction between monensin and growth promoters (GP). Monensin did not affect total digestibility (DEC) or relative liver size at 31 days of age. It significantly increased the relative length of the small intestine and decreased its specific gravity. AVO significantly improved digestibility at 31 days of age, but this was not observed at 53 days of age. BAC and VIR had no effect on this metric. In both age groups, AVO and VIR (but not BAC) reduced the size, length, and specific gravity of the small intestine, sometimes significantly. Our conclusion: BAC, VIR, and AVO cannot counteract the toxic effects of monensin. The performance-enhancing effects of growth hormone (GP) diminish or disappear with age, but its effect on reducing small intestine size remains significant in 49-day-old chicks. Non-human toxicity values Rats oral LD50 100 mg/kg Rats intraperitoneal LD50 15 mg/kg Mice oral LD50 43,800 μg/kg Mice intraperitoneal LD50 10 mg/kg Horses oral LD50 2 mg/kg |
| References |
|
| Additional Infomation |
Monensin is an antiprobiotic produced by Streptomyces cinnamonensis. It functions within the intestinal epithelial cells during the development of first-generation trophozoites into first-generation schizonts. It does not interfere with the host's acquired immunity against most coccidia. Monensin is a sodium and proton-selective ionocarrier and is therefore widely used in biochemical research.
See also: Monensin Sodium (note moved here). Monensin A is a spiroacetate, the main component of monensin, an antibiotic mixture produced by Streptomyces cinnamonensis. As an antiprobiotic, it is used in sodium salt form as a feed additive to prevent coccidiosis in poultry and as a growth promoter in cattle. It has dual action as an anticoccidial, antifungal, and ionocarrier. It is a monocarboxylic acid, cyclic hemiacetate, spiroacetate, and polyether antibiotic. Monensin is an antibacterial polyether isolated from Streptomyces cinnamonensis. It is widely used in ruminant feed. Monensin has also been reported to exist in Streptomyces glaucescens and Apis cerana, with relevant data available. Monensin is an antimicrobial agent produced by Streptomyces cinnamonensis. It functions within intestinal epithelial cells during the development of first-generation trophozoites into first-generation schizonts. It does not interfere with the host's acquired immunity against most coccidia. Monensin is a sodium and proton-selective ionoporter and is therefore widely used in biochemical research. See also: Monensin sodium (in salt form); bacitracin; monensin (ingredient). Avilamycin; Monensin (ingredients)...See more... Drug Indications For reducing the incidence of peripartum ketosis in dairy cows/growing cattle expected to develop ketosis. Mechanism of Action The Wnt signaling pathway is essential for embryonic development and maintaining adult tissue homeostasis. However, aberrant activation of this pathway is associated with a variety of human diseases, including gastrointestinal cancers, breast cancer, liver cancer, melanoma, and hematologic malignancies. In this study, we found that monensin (a polyether ionophore antibiotic) is a potent inhibitor of the Wnt signaling pathway. Inhibition of the Wnt/β-catenin signaling cascade by monensin was observed in mammalian cells stimulated with Wnt ligands, glycogen synthase kinase-3 inhibitors, and in cells transfected with β-catenin expression vectors. Furthermore, monensin inhibited Wnt-dependent caudal fin regeneration in zebrafish and Wnt or β-catenin-induced second body axis formation in Xenopus embryos. In Wnt3a-activated HEK293 cells, monensin blocked the phosphorylation of Wnt co-receptor LDL receptor-associated protein 6 and promoted its degradation. In human colorectal cancer cells with Wnt signaling pathway dysregulation, monensin reduced intracellular β-catenin levels. This reduction attenuated the expression of Wnt signaling pathway target genes, such as cyclin D1 and SP5, and decreased cell proliferation rate. In a mouse model of multiple intestinal tumors (Min), daily administration of monensin inhibited the progression of intestinal tumors without any toxic effects on normal mucosa. Our data suggest that monensin holds promise as a potential anticancer drug for treating tumors associated with Wnt signaling pathway dysregulation. Pre-incubation of rat soleus muscle with 1 μM and 10 μM monensin for 2 hours increased basal 2-deoxyglucose uptake by 76% and 121%, respectively. Under the same conditions, monensin reduced insulin (1 mU/mL)-stimulated 2-deoxyglucose uptake by 29% and 37%, respectively. Cytochalasin B inhibited monensin-induced basal 2-deoxyglucose uptake by 92%, indicating that this uptake process is mediated by glucose transporters. Monensin did not increase the accumulation of L-glucose in muscle cells, indicating that it does not affect cell membrane integrity. The stimulatory effect of monensin on basal 2-deoxyglucose uptake and its inhibitory effect on insulin-stimulated 2-deoxyglucose uptake were not affected by Ca2+ removal in the culture medium or by the sarcoplasmic reticulum Ca2+ release inhibitor dantrolene, suggesting that the effect of monensin is not mediated by calcium. Monensin had no effect on muscle ATP concentration. The monensin-induced increase in basal 2-deoxyglucose uptake was neither related to stimulation of muscle phosphatidylinositol 3-kinase activity nor inhibited by wollamedin, indicating that the increase in basal 2-deoxyglucose uptake is not mediated by activation of phosphatidylinositol 3-kinase. Monensin's inhibition of insulin-stimulated 2-deoxyglucose uptake was associated with a 31% decrease in insulin receptor abundance in muscle, a 64% decrease in insulin-induced insulin receptor β-subunit autophosphorylation, and a 44% decrease in insulin-stimulated phosphatidylinositol 3-kinase activity. The addition of monensin to the phosphatidylinositol 3-kinase response did not affect the enzyme's activity, indicating that the inhibition of this enzyme in monensin-treated muscle is indirect and occurs upstream of phosphatidylinositol 3-kinase. Therefore, it can be concluded that monensin has a dual effect on 2-deoxyglucose uptake in skeletal muscle: it stimulates basal uptake but inhibits insulin-stimulated uptake. The main reason for monensin's inhibition of insulin-stimulated uptake is its action on insulin receptors. |
| Molecular Formula |
C36H61O11-.NA+
|
|---|---|
| Molecular Weight |
692.85274
|
| Exact Mass |
692.411
|
| Elemental Analysis |
C, 62.41; H, 8.87; Na, 3.32; O, 25.40
|
| CAS # |
22373-78-0
|
| Related CAS # |
Monensin;17090-79-8; 22373-78-0 (sodium)
|
| PubChem CID |
23667299
|
| Appearance |
White to off-white solid powder
|
| Boiling Point |
766.3ºC at 760 mmHg
|
| Melting Point |
267-269ºC
|
| Flash Point |
229.2ºC
|
| Vapour Pressure |
4.13E-27mmHg at 25°C
|
| LogP |
2.873
|
| Hydrogen Bond Donor Count |
3
|
| Hydrogen Bond Acceptor Count |
11
|
| Rotatable Bond Count |
10
|
| Heavy Atom Count |
48
|
| Complexity |
1110
|
| Defined Atom Stereocenter Count |
17
|
| SMILES |
CC[C@]1(CC[C@@H](O1)[C@@]2(CC[C@@]3(O2)C[C@@H]([C@H]([C@H](O3)[C@@H](C)[C@H]([C@H](C)C(=O)[O-])OC)C)O)C)[C@H]4[C@H](C[C@@H](O4)[C@@H]5[C@H](C[C@H]([C@@](O5)(CO)O)C)C)C.[Na+]
|
| InChi Key |
XOIQMTLWECTKJL-FBZUZRIGSA-M
|
| InChi Code |
InChI=1S/C36H62O11.Na/c1-10-34(31-20(3)16-26(43-31)28-19(2)15-21(4)36(41,18-37)46-28)12-11-27(44-34)33(8)13-14-35(47-33)17-25(38)22(5)30(45-35)23(6)29(42-9)24(7)32(39)40;/h19-31,37-38,41H,10-18H2,1-9H3,(H,39,40);/q;+1/p-1/t19-,20-,21+,22+,23-,24-,25-,26+,27+,28-,29+,30-,31+,33-,34-,35+,36-;/m0./s1
|
| Chemical Name |
sodium;(2S,3R,4S)-4-[(2S,5R,7S,8R,9S)-2-[(2R,5S)-5-ethyl-5-[(2R,3S,5R)-5-[(2S,3S,5R,6R)-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyloxan-2-yl]-3-methyloxolan-2-yl]oxolan-2-yl]-7-hydroxy-2,8-dimethyl-1,10-dioxaspiro[4.5]decan-9-yl]-3-methoxy-2-methylpentanoate
|
| Synonyms |
Monensin sodium salt; Monensin sodium; 22373-78-0; DTXSID2045573; Monensin A sodium salt; sodium;(2S,3R,4S)-4-[(2S,5R,7S,8R,9S)-2-[(2R,5S)-5-ethyl-5-[(2R,3S,5R)-5-[(2S,3S,5R,6R)-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyloxan-2-yl]-3-methyloxolan-2-yl]oxolan-2-yl]-7-hydroxy-2,8-dimethyl-1,10-dioxaspiro[4.5]decan-9-yl]-3-methoxy-2-methylpentanoate; DTXCID0025573; C36H61NaO11;
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
Ethanol : ~20 mg/mL (~28.87 mM)
DMSO : ~5.4 mg/mL (~7.79 mM) |
|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2 mg/mL (2.89 mM) (saturation unknown) in 10% EtOH + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear EtOH + stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.4433 mL | 7.2166 mL | 14.4331 mL | |
| 5 mM | 0.2887 mL | 1.4433 mL | 2.8866 mL | |
| 10 mM | 0.1443 mL | 0.7217 mL | 1.4433 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved





























 COA
COA