| Size | Price | Stock | Qty |
|---|---|---|---|
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg | |||
| Other Sizes |
































| Targets |
DHODH
|
|---|---|
| ln Vitro |
Next, to address whether LAP/Lapachol inhibits the DHODH activity, we carried out a cell-free DHODH activity assay by measuring the reduction of DCIP. The hDHODH activity was significantly inhibited by LAP sodium salt with an IC50 value of 0.13 μM (Fig. 1c), indicating that LAP is a potent inhibitor of the hDHODH activity. [1]
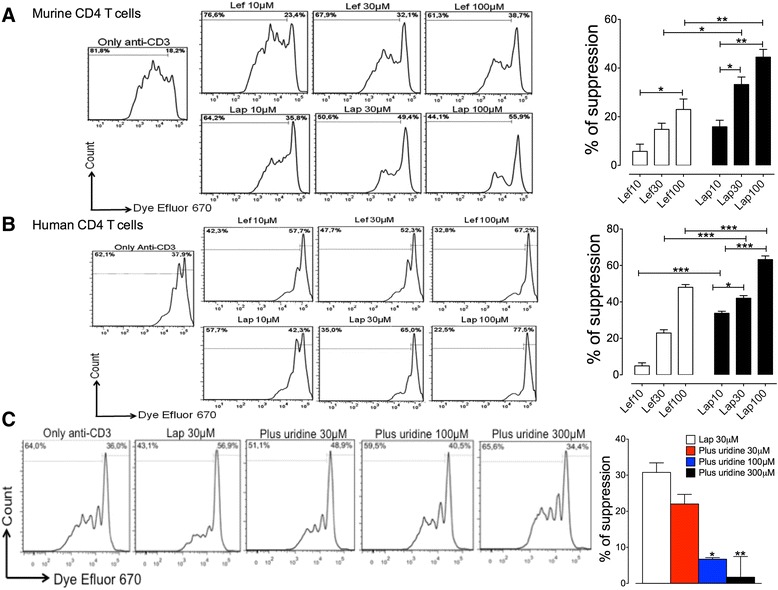
LAP/Lapachol inhibits lymphocyte proliferation through inhibition of pyrimidine biosynthesis [1] We next assessed the antiproliferative effect of LAP. To this end, freshly isolated mouse CD4 T cells were labelled with Dye Efluor 670 and stimulated with anti-CD3/CD28 in the presence of LAP or LEF (10, 30, and 100 μM) for 4 days. As shown in Fig. 2a, LAP or LEF inhibited the proliferation of murine CD4 T cells in a dose-dependent manner. We also investigated the effect of LAP in human CD4 T cells isolated from the peripheral blood of healthy donors. Similar to that observed with murine cells, we also found a dose-dependent inhibition of human T-cell proliferation in the presence of LAP or LEF (Fig. 2b). However, we found that LAP exhibited a greater ability to suppress the proliferation of human and murine CD4 T cells than was observed with LEF at the same equivalent concentrations (Fig. 2a and b). Additionally, we performed annexin-V/propidium iodide (PI) staining of human CD4 T cells treated with LAP or LEF to assess apoptotic cell death. While LEF showed no toxic effects at all concentrations, flow cytometric analysis revealed that only the highest concentration of LAP (100 μM) was toxic (Additional file 5: Table S3). Thus, the reduction of T-cell proliferation by LAP below 100 μM was primarily due to inhibition of the proliferative response rather than a reduction of cellular viability by toxicity. The antiproliferative effect of LEF is completely reversed by supplementation of uridine, supporting that DHODH is the target for LEF. We then investigated whether the antiproliferative effect of Lapachol/LAP is also due to targeting DHODH. To this end, human CD4 T cells were pretreated with LAP in the presence of different concentrations of uridine. As shown in Fig. 2c, uridine was able to reverse the antiproliferative effect of LAP in a dose-dependent manner. Of note, uridine alone had a minimal effect on the proliferative response of anti-CD3-stimulated T cells. Effect of oral administration of Lapachol on the experimental metastasis of B16BL6 melanoma cells [2] Lapachol was administered orally to mice three times (at − 48, − 24, and − 6 h) before tumor cell injection into the lateral tail vain. Two weeks after tumor cell injection, mice were euthanized, and the numbers of metastatic nodules in the lungs were counted. Low doses (0.5, 5, 10, and 20 mg/kg) of lapachol weakly but significantly inhibited metastasis, whereas higher doses (80 and 100 mg/kg) drastically promoted metastasis (Fig. 1A). To confirm the metastasis-inhibiting effect of low-dose lapachol, we increased the frequency of lapachol administration at 0.5 and 5.0 mg/kg. Although lapachol showed a tendency to inhibit metastasis, the inhibition did not increase with increased frequency of administration (Fig. 1B). In contrast to the weak inhibition of metastasis, the promotion of metastasis was substantial. Because the strong promotion of metastasis by a therapeutic agent calls for great concern, we further examined the metastasis-promoting property of Lapachol. To determine the sensitive period for metastasis promotion by lapachol, lapachol (80 mg/kg) was administered once at 48, 24, or 6 h before tumor cell injection. Only the administration at 6 h before tumor cell injection significantly increased the number of metastatic nodules in the lung (Fig. 1C). These results indicate that lapachol promotes metastasis within 6 h of administration, and this effect disappears within 24 h. Effect of in vitro treatment with Lapachol on the experimental metastasis of B16BL6 melanoma cells [2] To examine whether oral administration of lapachol promotes metastasis by directly affecting tumor cells or host factors, B16BL6 melanoma cells were treated with lapachol in vitro for 24 h and then injected iv into mice. The plasma concentration of lapachol in humans after a single oral dose of 35–40 mg/kg has been reported to be 5.6–26.4 μg/ml within 24 h (Block et al., 1974). We examined the effect of lapachol on B16BL6 melanoma cells in culture and found that treatment for 24 h with > 25 μg/ml lapachol completely killed B16BL6 melanoma cells (data not shown). In vitro treatment with lapachol at 20 μg/ml slightly enhanced the number of metastatic nodules in the lung (Fig. 1D), but this increase was much less than that observed after oral administration of lapachol (Fig. 1A and C). These results indicate that the promotion of metastasis by lapachol is largely derived from effects on host factors and not from direct effects on tumor cells. Low cytotoxicity in HepG2 cells (3405.8 ± 261.33 μM), good anti-Leishmania activity, and favorable selectivity indexes (SI) against promastigotes of both L. amazonensis (IC50 = 79.84 ± 9.10 μM, SI = 42.65) and L. infantum (IC50 = 135.79 ± 33.04 μM, SI = 25.08) were observed. Furthermore, anti-Leishmania activity assays performed on intracellular amastigotes showed good activity for Lapacholl (IC50 = 191.95 μM for L. amazonensis and 171.26 μM for L. infantum). Flow cytometric analysis demonstrated that the cytotoxic effect of lapachol in Leishmania promastigotes was caused by apoptosis-like death [3]. |
| ln Vivo |
In a CIA mouse model, lapacol (3, 10 mg/kg; wall gavage; once daily for 4 weeks) lessens the severity of experimental arthritis [1]. When used once daily for nine days, lapachol (10 mg/kg; wall gavage) significantly lowers the respiration of knee leukocytes in adult C57BL/6 mice that have disaster-induced arthritis (AIA) [1].
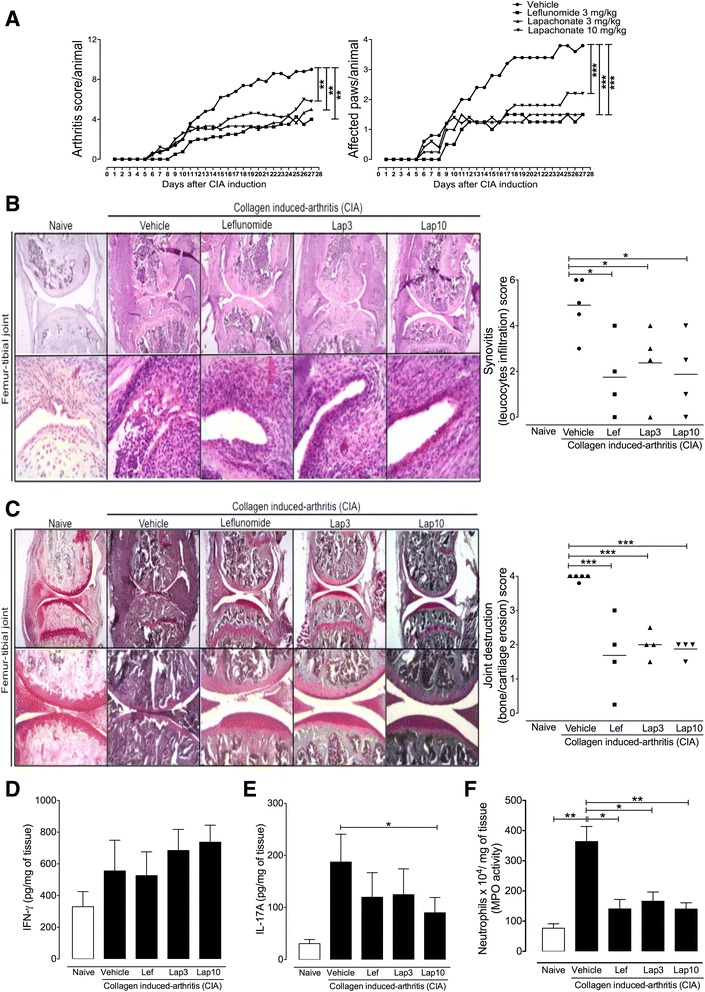
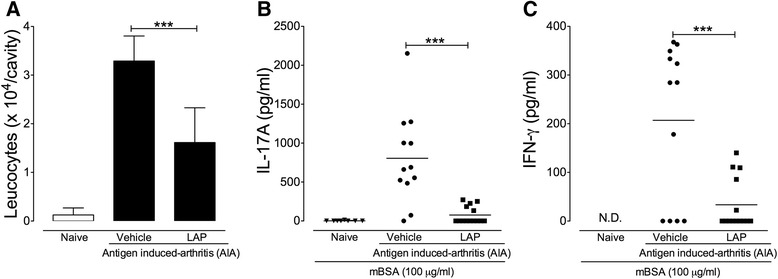
LAP/Lapachol reduces the severity of experimental arthritis [1] We next examined the therapeutic potential of LAP in two experimental models of arthritis. The first model was collagen-induced arthritis (CIA), a well-established T cell-dependent preclinical model for RA. Treatment with LAP was started just after booster injection with collagen on day 21 after the first immunization. Mice were orally treated with LAP (3 mg/kg and 10 mg/kg) once a day for 4 weeks. LAP was well tolerated without apparent side effects based on observations of the general symptoms of toxicity, including piloerection, diarrhea, weight loss, and prostration. We also found that treatment with LAP did not alter the serum levels of ALT or AST during the CIA protocol (Additional file 6: Figure S3), indicating that it was not hepatotoxic at the doses used. As a positive therapeutic control, mice were treated with LEF (3 mg/kg) using the same treatment schedule. The doses of LEF and LAP used in this therapeutic protocol were based on previous reports. Clinical arthritis scores were recorded from booster injection (day 0) and graded on a scale of the magnitude of paw swelling, erythema, and ankylosis (as described in the Methods section). We found that LAP, at both doses used, markedly attenuated the severity of arthritis in CIA mice, similar to those observed in mice treated with LEF, as evidenced by a reduction of clinical score and the number of affected paws (Fig. 3a). Histopathological analysis of knee joint sections from vehicle-treated mice stained with H&E and Safranin-O revealed inflammatory cell infiltration, pannus formation, and cartilage loss when compared to naive mice (Fig. 3b and c). Notably, LAP markedly reduced all histopathological features of arthritis severity when compared to the vehicle-treated group. No significant differences in histopathological features were observed between mice treated with LAP and LEF (Fig. 3b and c). Additionally, we measured the levels of inflammatory cytokines and MPO activity, which indirectly reflects neutrophil infiltration, in the hind paws from CIA mice treated or not with LAP or LEF. We did not found any significant differences in IFN-γ levels among all groups (Fig. 3d). However, CIA mice treated with LAP with the dose of 10 mg/kg showed a significant reduction in IL-17A levels (Fig. 3e). Moreover, we found that mice treated with LAP or LEF showed reduced MPO activity compared to vehicle-treated CIA mice (Fig. 3e). Finally, we investigated the immunomodulatory effects of LAP/Lapachol in a second model of experimental arthritis. To this end, we employed the antigen-induced arthritis (AIA) model in C57BL/6 mice, which also requires a T-cell response for the generation of the acute articular inflammation. Briefly, mice were treated orally with LAP (10 mg/kg) once a day over 9 days, beginning 12 days after the first immunization with the antigen mBSA. On day 21 after the first immunization, arthritis was induced by intra-articular injection of mBSA into the knees of immunized mice. We did not find differences in the serum levels of anti-mBSA total IgG between mBSA-immunized mice treated or not (vehicle) with LAP (Additional file 7: Figure S4). However, mice treated with LAP exhibited a remarkable reduction in leucocyte infiltration into the knee joint 6 h after mBSA challenge when compared to vehicle-treated mice (Fig. 4a). We then evaluated the recall responses by cells from mBSA-immunized mice treated or not with LAP. The mBSA-specific production of IL-17 and IFN-γ by draining lymph node cells and splenocytes was significantly reduced in mice treated with LAP (Fig. 4b and c). IL-4 was not detected in the supernatant of stimulated cells (data not shown). Collectively, these findings show the marked immunomodulatory effects of LAP in two models of experimental arthritis. Promotion of experimental metastasis of B16BL6 melanoma cells by oral administration of Lapachol in T-cell-deficient mice and NK-suppressed mice [2] Immune cells, such as T cells and NK cells, are postulated to be responsible for detecting and eliminating tumor cells (Trinchieri, 1989, Jakobisiak et al., 2003, Mehlen and Puisieux, 2006). To examine whether metastasis enhancement by lapachol is mediated by these immune cells, a metastasis assay was conducted using T-cell-deficient nude mice and NK-suppressed mice. When B16BL6 melanoma cells were injected iv into these mice 6 h after oral administration of lapachol, the number of metastatic nodules in the lung increased drastically (Fig. 2), as in normal mice (Fig. 1C). These observations indicate that lapachol can promote metastasis in the absence of T cells and NK cells. However, the possibility that lapachol affects T cells and NK cells cannot be excluded without examining the effects of lapachol on these immune cells in normal mice. Protein C activity and prothrombin time after oral administration of vitamin K antagonist [2] Vitamin K antagonists can transiently induce a hypercoagulable state upon initiation of treatment (Vigano et al., 1984, Stirling, 1995, Srinivasan et al., 2004). A hypercoagulable state promotes blood-borne metastasis in cancer patients (Bick, 1992, Rickles et al., 1992, Mousa et al., 2006) and in experimental animals (Cliffton et al., 1961, Cliffton and Agostino, 1964). To examine whether hypercoagulability induced by Lapachol might promote metastasis, protein C activity and the prothrombin time were measured after oral administration of lapachol. Protein C activity declined significantly within 3 h of lapachol administration, was reduced maximally between 6 and 12 h, and recovered slowly between 12 and 24 h. On the other hand, the prothrombin time was prolonged significantly only at 12 h after lapachol administration (Fig. 4A). Six hours after lapachol administration, protein C activity was maximally suppressed, but the prothrombin time was not prolonged. Therefore, the hypercoagulable state occurred at least 6 h after lapachol administration. These time courses for the hypercoagulable state were consistent with the observations of increased metastasis when tumor cells were injected 6 h after lapachol administration and of a lack of metastasis promotion when tumor cells were injected > 24 h after lapachol administration (Fig. 1B). To evaluate the effect of Leishmania on host tissues, murine models of CL and VL were submitted to a therapeutic protocol using orally administered Lapachol. The animals were euthanized after the treatment. No significant differences in lesion size and macroscopic changes in liver or spleen were observed during sample collection (data not shown). The treatment of CL with lapachol or Amb significantly reduced parasite load in mice skin compared to the control group (negative treatment group) (p < 0.05). The mean number of parasites in the skin lesion (5.4 × 108 parasites/mg) determined for the lapachol group was approximately 24.5 times lower than that in the non-treated group (1.324 × 1010 parasites/mg). Similarly, animals treated with Amb (positive treatment group) showed 33.5 times fewer parasites (3.95 × 108) per milligram of skin compared to the non-treated control group (Fig. 4B). [3] Another study demonstrated that oral application of Lapachol in hamsters infected with L. braziliensis did not significantly reduce the parasite load in the lesion (Teixeira et al., 2001). Although lapachol was administered orally in this study, the model of CL (BALB/c mice instead hamsters), Leishmania species, therapeutic protocol, and methodology for evaluating parasitic load were different from that of the previous study, which might explain the differences in the results. [3] Strikingly, Lapachol treatment of mice with VL significantly reduced the parasitic load in the spleen and liver of the animals (p < 0.05). Mice treated with lapachol presented approximately 4.6 and 5.3 fewer parasites in the spleen and liver, respectively, compared to non-treated animals. When treated with Amb, mice presented approximately 18.6 and 54.4 lesser parasites in the spleen and liver respectively, compared to non-treated animals (Fig. 4A) [3]. |
| Enzyme Assay |
Enzymatic assay [1]
hDHODH activity was assessed using a colorimetric continuous assay that monitors 2,6-dichloroindophenol (DCIP) reduction. Change in absorbance at 610 nm was monitored over a period of 60 s at 25 °C using a microplate reader. The enzymatic reaction was analyzed in a total volume of 195 μl containing 50 mmol/l Tris, pH 8.15, 150 mmol/l KCl, 0.1% Triton X-100, 1 mmol/l l-dihydroorotate, 100 μmol/l CoQ0, and 60 μmol/l DCIP. The assay was started with 5 μl of 0.8 μmol/l stock of enzyme prepared in 50 mmol/l HEPES, pH 7.7, 400 mmol/l NaCl, 10% glycerol, 0.05% Thesit, and 1 mmol/l EDTA in a final concentration of enzyme at 20 nmol/l. A reference measurement was obtained by preparing the same solution without enzyme. LAP/Lapachol was analyzed in quadruplicate for each concentration used. LAP/Lapachol sodium salt was prepared as a 10 mmol/l stock in DMSO. From this solution, dilutions were prepared in the assay mixture to achieve the range of 100 μmol/l to 0.35 nmol/l. Control enzyme activity in the absence of inhibitor was taken as 100%. The percentage of activity versus log of LAP/Lapachol concentration graph was drawn. The half maximal inhibitory concentration (IC50) values were calculated using a nonlinear fitting of the concentration–response data to the equation. |
| Cell Assay |
Cell Proliferation Assay[1]
Cell Types: Murine CD4 T cells Tested Concentrations: 10, 30, 100 μM Incubation Duration: 4 days Experimental Results: Inhibited the proliferation of mouse CD4 T cells in a dose-dependent manner. Effect of Lapachol on promastigote viability in vitro [3] The inhibitory concentrations (IC50) of lapachol against promastigotes of L. infantum and L. amazonensis were determined using the resazurin-based colorimetric assay (Corral et al., 2013). Log-phase promastigotes (2.5 × 105 parasites/well) were seeded in flat-bottom 96-well cell culture plates in complete α-MEM and incubated at 26 °C. Lapachol was two-fold serially diluted over seven concentrations (from 412 μM to 6 μM) in complete α-MEM and each concentration was tested in triplicate. Amb was used as a positive control (0.54–0.008 μM). Non-treated parasites were used for comparing viability. The cells were incubated with the substances for 48 h, after which, resazurin solution (10% v/v) was added to the wells and the plates were incubated for 4 h. Fluorescence (Spectramax M2, Molecular Devices LLC, USA) was measured at 550 nm excitation and 590 nm emission wavelength. Fluorescence intensity was expressed as arbitrary units. In vitro cytotoxicity of Lapachol toward the HepG2 cell line [3] The cytotoxicity concentration (CC50) of the substances against HepG2 cells was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT) assay (Mosmann, 1983). Cells were seeded in 96-well flat bottom plates at 5 × 104 cells/well in complete RPMI-1640 medium and maintained for 24 h at 37 °C in a humidified 5% CO2 atmosphere. Lapachol was serially diluted two-fold over seven concentrations (from 4.12 mM to 0.016 μM) and added to the plates, which were incubated for 48 h at 37 °C in the presence of 5% CO2. Subsequently, MTT solution (5 mg/mL, 50 μg/well) was added to the wells and the plates were incubated for additional 4 h. The supernatants were aspirated and the formazan crystals formed were dissolved in DMSO. Absorbance was determined in a spectrophotometer at 570 nm (Dutta et al., 2005, Mosmann, 1983). Non-treated HepG2 cells and Amb (1.080 mM–17 μM) were used as controls. Lapachol was tested both for leishmanicidal activity and cytotoxicity in technical triplicates on microplates and the results are representative of three independent experiments (biological triplicate). Lapachol was tested both for leishmanicidal activity and cytotoxicity in technical triplicates on microplates and the results are representative of three independent experiments (biological triplicate). Efficacy of Lapachol against intracellular amastigotes [3] Immortalized murine macrophages, RAW 264.7, cultured in complete RPMI-1640 medium and maintained at 37 °C in an atmosphere of 5% CO2 and 95% humidity, were seeded (5 × 104/well) on a 24-well tissue culture plate containing circular coverslips in each well and incubated (37 °C, 5% CO2) for 4 h to allow cell adherence. Then, Leishmania promastigotes in late stationary growth phase were added to interact with the macrophages in the proportion of 10 promastigotes/macrophage/well for 24 h. Subsequently, two-fold serially diluted lapachol (ranging from 660 μM to 41 μM) and Amb (ranging from 0.54 to 0.03 μM) were added to the wells. After 48 h, the coverslips were removed and stained with rapid panoptic, mounted in glass slides using Canada balsam, and analyzed using light microscopy to determine the infection rate of macrophages. The values obtained for each concentration [(number of infected macrophages/300 counted macrophages) × 100] were used to obtain the amastigote intracellular IC50 value for lapachol. The IC50 for the effect of lapachol on the intracellular amastigotes was expressed as the concentration necessary to halve the number of the infected macrophages compared to non-treated control cells (Vermeersch et al., 2009). |
| Animal Protocol |
Animal/Disease Models: Male DBA1/J mice (10-12 weeks old) collagen-induced arthritis (CIA) model [1]
Doses: 3 mg/kg and 10 mg/kg Route of Administration: Oral; one time/day for 4 weeks Experimental Results: Both doses Dramatically diminished the severity of arthritis in CIA mice. Dramatically diminished all histopathological features of arthritis severity. Collagen-induced arthritis (CIA) [1] Male DBA/1 J mice were injected i.d. at the base of the tail with 200 μg bovine type II collagen (CII) emulsified in Freund’s complete adjuvant (CFA) on day 0. Mice were boosted i.d. with CII (200 μg emulsified in Freund’s incomplete adjuvant (IFA)) on day 21. Mice were monitored daily for signs of arthritis. Scores were assigned based on erythema, swelling, or ankylosis present in each paw on a scale of 0 to 3, giving a maximum score of 12 per mouse. After arthritis induction, mice were treated orally with LAP/Lapachol (3 mg/kg and 10 mg/kg) or LEF (3 mg/kg) or saline daily. The clinical score was addressed every day after collagen boost. All mice were euthanized for histologic assessment of the hind limbs 4 weeks after the boost. Antigen-induced arthritis (AIA) [1] Mice were immunized with methylated bovine serum albumin as described previously. Briefly, mice were immunized with subcutaneous injection of an emulsion with mBSA (500 μg) and CFA (2 mg/ml of inactivated Mycobacterium tuberculosis). Booster injections of mBSA in IFA were given at 7 and 14 days after the first immunization. On day 21 after the first immunization, arthritis was induced by an intra-articular injection of mBSA (30 μg). During the AIA protocol, LAP/Lapachol (10 mg/kg) or saline (vehicle) was given orally every day from 12 to 21 days after first immunization. Drug administration and treatment [2] Lapachol was dissolved in 2% sodium carbonate in water, and warfarin was dissolved in water. These drugs were administered by oral gavage. Vitamin K3 was dissolved in sterile phosphate-buffered saline (PBS) and administered ip. The solutions were freshly prepared just before use. For in vitro treatment, Lapachol was dissolved in ethanol at a concentration of 40 mM and added to cultures. Ethanol was added to the control culture at a concentration of 0.05%. In vivo efficacy of Lapachol against leishmaniasis [3] The present study was approved by the Ethical Committee for Animal Experimentation of the Federal University of Uberlândia (protocol number 069/2013), and all procedures were performed according to the international guidelines (Principles of Laboratory Animal Care (1985)). Cutaneous leishmaniasis model [3] Eighteen BALB/c mice (8-week-old females) were infected with 1 × 107 metacyclic promastigotes of L. amazonensis in the tail base via subcutaneous route. The mice were randomly divided into three groups 30 days after infection: (a) Lapachol (n = 6; 25 mg/kg for 24 h oral route for 10 days), (b) Amb (n = 6; 5 mg/kg for 24 h; intraperitoneal route for 10 days), and (c) PBS (n = 6; oral route for 24 h over 10 days). The animals were euthanized after the treatment and skin lesions were collected and used for determining parasitic load using quantitative polymerase chain reaction (qPCR). Visceral leishmaniasis model [3] Eighteen BALB/c mice (8-week-old females) were infected with 1 × 107 metacyclic promastigotes of L. infantum via the intraperitoneal route. Twenty days after infection, mice were randomly divided into three groups: (a) Lapachol (n = 6; 25 mg/kg for 24 h oral route for 10 days), (b) amphotericin B (n = 6; 5 mg/kg for 24 h; intraperitoneal route for 10 days), and (c) PBS (n = 6; oral route for 24 h over 10 days). |
| ADME/Pharmacokinetics |
The stability of lapasol sodium in plasma and acidic media (similar to the gastric environment) was analyzed using ultra-high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). Results confirmed that lapasol sodium can be instantaneously converted to neutral lapasol molecules. After intravenous administration, the plasma concentration profiles of lapasol and lapasol sodium were completely consistent, conforming to a two-compartment open model, with a volume of distribution of 0.19 ± 0.03 L/kg, a total clearance of 0.04 ± 0.01 L/h/kg, and a half-life of 4.1 ± 1.1 h (Supplementary Document 1: Figure S1; Supplementary Document 2: Table S1). Linear pharmacokinetic characteristics were observed within the studied dose range (2–25 mg/kg). The plasma concentration profiles after oral administration of LAP and LAP sodium (two different doses) best conformed to a one-compartment model, with bioavailabilities of 55–77% and 42%, respectively (Supplementary Document 1: Figure S1; Supplementary Document 3: Table S2). [1]
Preparation of LAP sodium salt[1] Dissolve LAP (500 mg, 2.06 mmol) in ethanol (20 ml), add NaOH (112 mg, 0.28 mmol), and stir the reaction mixture for 24 hours. After LAP consumption, the reaction mixture was concentrated under reduced pressure. The solid residue was washed with dichloromethane (4 times) and petroleum ether (4 times) to give 518 mg of purple solid, with a yield of 95% (¹H NMR (300 MHz, D₂Od₆) δ 7.66 (br s, 1H), 7.64 (br s, 1H), 7.53 (br t, J = 9 Hz, 1H), 7.41 (br t, J = 9 Hz, 1H), 5.15 (br t, J = 9 Hz, 1H), 3.06 (d, J = 6 Hz, 2H), 1.72 (s, 3H), 1.63 (s, 3H); ¹³C NMR (101 MHz, DMSOd₆) δ 187.2, 178.4, 169.6, 1000 ppm). 136.0, 133.2, 131.3, 129.7, 127.5, 125.76, 124.81, 124.4, 117.9, 25.59, 20.8, 14.1; HRMS-ESI m/z calculated value: [M + Na]+ = 265.0835; measured value = 265.0834). Pharmacokinetic Study Design[1] To administer to Wistar rats, LAP/lapamil was dissolved in DMSO:Tween 80:5% glucose solution at a volume ratio of 15:5:80 to obtain solutions with concentrations of 1 mg/ml (for intravenous injection) and 5 mg/ml (for oral administration). Rats were given LAP via intravenous bolus at doses of 2 mg/kg (n = 7) and oral doses of 10 mg/kg (n = 8) and 25 mg/kg (n = 6), respectively. LAP salts were administered via intravenous injection (2 mg/kg, n = 6) and oral administration (30 mg/kg, equivalent to 27.5 mg/kg of LAP, n = 8). Intravenous doses were administered via caudal vein, and oral doses were administered via gavage. Dosage selection was based on prior toxicological and pharmacodynamic studies [20]. Blood samples (200–250 μl) were drawn into heparinized tubes via puncture of the caudal vein (opposite to the administration vein) at predetermined time points (30 minutes before administration and 0.08, 0.25, 0.5, 1, 2, 4, 6, 8, 12, and 24 hours after administration). The same method was used after oral administration of LAP, and blood samples were collected at 0.25, 0.5, 1, 1.5, 2, 3, 6, 12, 24 and 30 hours. Blood samples were collected 10 hours and 12 hours after intravenous injection and oral administration of LAP sodium, respectively. Blood samples were separated by centrifugation (6800 × g, 4 °C, 10 min) to obtain plasma, which was stored at –80 °C until UPLC-MS/MS analysis. Pharmacokinetic analysis [1] After intravenous and oral administration, the pharmacokinetic parameters of lapasol/Lapachol and lapasol sodium were determined by analyzing individual plasma concentration-time curves using a non-compartmental model (NCA). After oral administration, the peak plasma concentration (Cmax) and time to peak (Tmax) were obtained by visually observing the plasma concentration-time curve data. Elimination rate constant (λ), area under the curve (AUC0–∞), clearance (CLtot), half-life (t1/2), volume of distribution (Vdss), mean residence time (MRT), and bioavailability (Fabs) were calculated using classical equations. Compartmental model analysis was performed using SCIENTIST v.2.0.1 software. One-compartment and two-compartment models with or without weighting schemes were evaluated. The model best suited to the data was selected based on the random distribution of residuals, correlation coefficients, and the model selection criteria (MSC) provided by the software. Individual plasma concentration curves of LAP/lapamol and LAP sodium after intravenous injection best fit the two-compartment open model. Plasma concentration curves after oral administration of two different doses of LAP and one dose of LAP sodium best fit the one-compartment model. Plasma analysis was performed using UPLC-MS/MS [1]. The concentration of LAP/lapamol in plasma samples was determined using a validated (FDA, 2001) UPLC-MS/MS method. Analysis was performed on an Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm particle size) at a flow rate of 300 μL/min and a column temperature of 35 °C. The mobile phase was water (A) and acetonitrile (B), acidified with 0.1% acetic acid, using a gradient elution program as follows: 0 min (90% A), 1 min (75% A), 7 min (50% A), 8.5 min (0% A), 9.5 min (100% A). The triple quadrupole mass spectrometer parameters were set as follows: capillary voltage (2.20 kV); extractor voltage (3.0 V); ion source temperature (150 °C); desolventizing temperature (300 °C); cone gas flow rate (50 L/h); desolventizing gas flow rate (700 L/h). Quantitative analysis was performed using multiple reaction monitoring (MRM). For LAP, using a cone energy of 24 V and a collision energy of 19 V, the transition with m/z 243 > 187 was determined to be most suitable for quantitative analysis (the calibration curve for LAP is for concentrations ranging from 1 to 20,000 ng/ml, R > 0.99, with a limit of quantitation of 1 ng/ml and a limit of detection of 0.1 ng/ml). Sample preparation for pharmacokinetic studies [1] 200 μl of cold acetonitrile containing 5 μg/ml internal standard (2-methylaminolapamol) and 0.05% trifluoroacetic acid was added to 100 μl of plasma and vortexed for 20 seconds. The precipitated protein was removed by centrifugation (4 °C, 6800 × g, 10 min). 200 μl of the supernatant was diluted 1:1 with pure water, filtered through a 0.22 μm filter membrane, and then analyzed. To prepare the calibration curve, LAP/lapamol was added to a blank plasma sample and subsequent processing was performed as instructed. Animal samples with concentrations at the upper limit of the calibration curve were diluted with blank plasma before processing. |
| Toxicity/Toxicokinetics |
3884 rat oral LDLo 1200 mg/kg, Toxicology and Applied Pharmacology, 17(1), 1970 [PMID:4989601]
3884 mouse oral LD50 487 mg/kg Behavior: somnolence (overall activity inhibition); Gastrointestinal tract: hypermotility, diarrhea; Skin and appendages (skin): dermatitis; Other: post-systemic exposure, Toxicology and Applied Pharmacology, 17(1), 1970 [PMID:4989601] 3884 mouse intraperitoneal injection LD50 400 mg/kg, Journal of Pharmaceutical Chemistry, 26(570), 1983 |
| References |
[1]. Lapachol, a compound targeting pyrimidine metabolism, ameliorates experimental autoimmune arthritis. Arthritis Res Ther. 2017 Mar 7;19(1):47.
[2]. Promotion or suppression of experimental metastasis of B16 melanoma cells after oral administration of lapachol. Toxicol Appl Pharmacol. 2008 Jun 1;229(2):232-8. [3]. Efficacy of lapachol on treatment of cutaneous and visceral leishmaniasis. Exp Parasitol. 2019 Apr:199:67-73. |
| Additional Infomation |
Lapachol is a hydroxy-1,4-naphthoquinone, where 1,4-naphthoquinone is substituted at the 2 and 3 positions with a hydroxyl group and a 3-methylbut-2-en-1-yl group, respectively. It is a natural compound with antibacterial and anticancer properties, first isolated from the bark of Tabebuia avellanedae, a plant in the Bignoniaceae family, in 1882. It functions as a plant metabolite, antitumor agent, antibacterial agent, and anti-inflammatory agent. It is a hydroxy-1,4-naphthoquinone olefin compound. Lapachol has been reported to exist in Catalpa ovata, Plenckia populnea, and other organisms with relevant data. Background: Inhibiting pyrimidine biosynthesis and blocking the activity of dihydroorotate dehydrogenase (DHODH) (the main target of leflunomide) has proven to be an effective strategy for treating rheumatoid arthritis (RA). However, a significant proportion of RA patients do not respond to LEF treatment. This study investigated the immunosuppressive properties of lapasol (LAP), a natural naphthoquinone compound, as a potential DHODH inhibitor. Methods: We performed molecular flexible docking studies and bioactivity assays to determine the ability of LAP to interact with and inhibit DHODH activity. Furthermore, we conducted in vitro studies using isolated lymphocytes to evaluate the antiproliferative effect of LAP. Finally, we used collagen-induced arthritis (CIA) and antigen-induced arthritis (AIA) models to investigate the anti-arthritis effect of LAP. Results: We found that LAP is a potent DHODH inhibitor with a significant ability to inhibit the proliferation of human and mouse lymphocytes in vitro. Importantly, uridine supplementation eliminated the antiproliferative effect of LAP, indicating that the pyrimidine metabolic pathway is the target of LAP. In vivo experiments showed that LAP treatment significantly reduced the progression of CIA and AIA, as evidenced by decreased clinical scores, joint tissue damage, and inflammation. Conclusion: Our findings present a binding model and confirm that LAP can inhibit DHODH, thereby reducing lymphocyte proliferation and alleviating the severity of experimental autoimmune arthritis. Therefore, LAP can be considered a potential immunosuppressant with potential application value in the treatment of rheumatoid arthritis. [1] Lapazol [2-hydroxy-3-(3-methyl-2-butenyl)-1,4-naphthoquinone] is a vitamin K antagonist with antitumor activity. This study investigated the effect of lapazol on experimental metastasis of mouse B16BL6 melanoma cells. A single oral administration of a high-toxicity dose of lapazol (80-100 mg/kg) 6 hours before intravenous injection of tumor cells significantly promoted tumor metastasis. This metastasis-promoting effect was also observed in T-cell deficient mice and NK-cell suppressed mice. In vitro treatment of B16BL6 cells with lapazol only slightly promoted metastasis, indicating that the main mechanism by which lapazol promotes metastasis is by affecting host factors other than T cells and NK cells. Six hours before intravenous injection of tumor cells, a single oral dose of warfarin, the most commonly used vitamin K antagonist, also significantly promoted the metastasis of B16BL6 cells. Pre-administration of vitamin K3 almost completely inhibited the metastatic effects of lapasol and warfarin, suggesting that the metastatic effect of lapasol is due to vitamin K antagonism. Six hours after oral administration of lapasol or warfarin, protein C levels dropped to their lowest levels, while prothrombin time was not prolonged. These observations suggest that high-toxic doses of lapasol promote metastasis by inducing a hypercoagulable state through inhibition of the vitamin K-dependent pathway. On the other hand, continuous oral administration of low doses (5-20 mg/kg) of lapasol can weakly but significantly inhibit metastasis, the mechanism of which is unclear, suggesting that lapasol may have the potential to be an anti-metastatic drug. [2] Leishmaniasis is one of the most important neglected diseases worldwide. It is a life-threatening disease that leads to a severe disease burden, long-term disability and premature death. Treatment options include disease control or interventions, but currently available drugs require long-term treatment and have issues with toxicity and reduced efficacy. Natural products isolated from plants, such as lapachol, an abundant naphthoquinone compound naturally found in plants of the genus Tabebuia (Bignoniaceae) in South America, are a promising option for the treatment of leishmaniasis. This study investigated the in vitro and in vivo killing activity of lapachol against Leishmania infantum and L. amazonensis, pathogens of visceral and cutaneous leishmaniasis, respectively. Low cytotoxicity (3405.8 ± 261.33 μM), good anti-leishmaniatic activity, and good selectivity index (SI) against L. amazonensis (IC50 = 79.84 ± 9.10 μM, SI = 42.65) and proflagellates of L. infantum (IC50 = 135.79 ± 33.04 μM, SI = 25.08) were observed in HepG2 cells. In addition, the assay of anti-leishmaniasis activity against intracellular aflagellates showed that lapasol had good activity (IC50 = 191.95 μM against Leishmania javanica and IC50 = 171.26 μM against Leishmania infantis). Flow cytometry analysis showed that the cytotoxic effect of lapasol against Leishmania proflagellates was caused by apoptosis-like death. Interestingly, the in vitro leishmanic effect of lapasol was validated in vivo in mouse visceral and cutaneous leishmaniasis models. Lapasol (25 mg/kg, orally, over 24 hours for 10 days) significantly reduced the parasite load in skin lesions, liver and spleen, with effects similar to the reference drug amphotericin B. These results reinforce the therapeutic potential of lapasol and warrant further investigation as an anti-leishmaniasis treatment. [3]
|
| Molecular Formula |
C15H14O3
|
|---|---|
| Molecular Weight |
242.27
|
| Exact Mass |
242.094
|
| Elemental Analysis |
C, 74.36; H, 5.82; O, 19.81
|
| CAS # |
84-79-7
|
| PubChem CID |
3884
|
| Appearance |
Light yellow to yellow solid powder
|
| Density |
1.2±0.1 g/cm3
|
| Boiling Point |
390.1±42.0 °C at 760 mmHg
|
| Melting Point |
141-143ºC(lit.)
|
| Flash Point |
203.9±24.4 °C
|
| Vapour Pressure |
0.0±0.9 mmHg at 25°C
|
| Index of Refraction |
1.606
|
| LogP |
4.08
|
| Hydrogen Bond Donor Count |
1
|
| Hydrogen Bond Acceptor Count |
3
|
| Rotatable Bond Count |
2
|
| Heavy Atom Count |
18
|
| Complexity |
439
|
| Defined Atom Stereocenter Count |
0
|
| SMILES |
O=C1C2C(=CC=CC=2)C(=O)C(C/C=C(\C)/C)=C1O
|
| InChi Key |
CWPGNVFCJOPXFB-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C15H14O3/c1-9(2)7-8-12-13(16)10-5-3-4-6-11(10)14(17)15(12)18/h3-7,16H,8H2,1-2H3
|
| Chemical Name |
4-hydroxy-3-(3-methylbut-2-enyl)naphthalene-1,2-dione
|
| Synonyms |
NSC 11905; 84-79-7; 2-Hydroxy-3-(3-methylbut-2-enyl)-1,4-naphthoquinone; lapachol; Greenhartin; Bethabarra wood; Taiguic acid; Lapachol wood; Taigu wood; NSC-11905; Lapachol
|
| HS Tariff Code |
2934.99.9001
|
| Storage |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture. |
| Shipping Condition |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| Solubility (In Vitro) |
DMSO : ~66.67 mg/mL (~275.19 mM)
|
|---|---|
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (10.32 mM) (saturation unknown) in 10% DMSO + 40% PEG300 +5% Tween-80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution.
For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 + to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.1276 mL | 20.6381 mL | 41.2763 mL | |
| 5 mM | 0.8255 mL | 4.1276 mL | 8.2553 mL | |
| 10 mM | 0.4128 mL | 2.0638 mL | 4.1276 mL |
*Note: Please select an appropriate solvent for the preparation of stock solution based on your experiment needs. For most products, DMSO can be used for preparing stock solutions (e.g. 5 mM, 10 mM, or 20 mM concentration); some products with high aqueous solubility may be dissolved in water directly. Solubility information is available at the above Solubility Data section. Once the stock solution is prepared, aliquot it to routine usage volumes and store at -20°C or -80°C. Avoid repeated freeze and thaw cycles.
Calculation results
Working concentration: mg/mL;
Method for preparing DMSO stock solution: mg drug pre-dissolved in μL DMSO (stock solution concentration mg/mL). Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug.
Method for preparing in vivo formulation::Take μL DMSO stock solution, next add μL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O,mix and clarify.
(1) Please be sure that the solution is clear before the addition of next solvent. Dissolution methods like vortex, ultrasound or warming and heat may be used to aid dissolving.
(2) Be sure to add the solvent(s) in order.
|
|
|
Products are for research use only; We do not sell to patients
Copyright 2020 InvivoChem LLC | All Rights Reserved